Your new post is loading...

|
Scooped by
mhryu@live.com
February 6, 4:56 PM
|
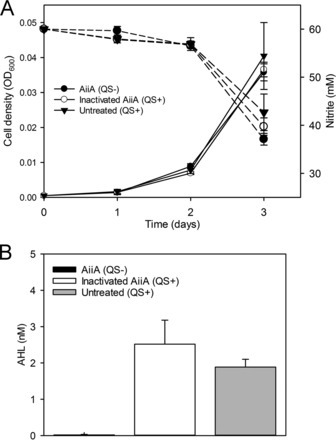
Unlike genome editing, RNA editing offers the ability to transiently alter cells with minimal risk from off-target effects. While exon-skipping technologies can influence splice site selection, many desired perturbations to the transcriptome require replacement or addition of exogenous exons to target mRNAs, such as replacing disease-causing exons, repairing truncated proteins, or engineering protein fusions. Here, we report the development of RNA-guided trans-splicing with Cas editor (RESPLICE). RESPLICE uses two orthogonal RNA-targeting CRISPR effectors to co-localize a trans-splicing pre-mRNA and to inhibit the cis-splicing reaction, respectively. We demonstrate efficient, specific, and programmable trans-splicing of RNA cargo (up to 2.1 kb) into 11 endogenous transcripts across 3 cell types, achieving up to 45% trans-splicing efficiency in bulk or 90% when sorting for high effector expression. Our results present RESPLICE as a mode of RNA editing that could provide fine-tuned and transient control of cellular programs.
|
|
Scooped by
mhryu@live.com
February 6, 4:43 PM
|
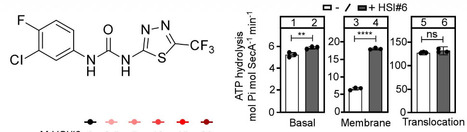
The Sec pathway is an essential protein secretion route for all organisms. In bacteria, the SecA ATPase peripherally associates with the SecYEG channel to form the translocase that mediates preprotein export. Activation of the translocase depends strictly on the synergy of signal peptide and mature domain binding. Thus, client selectivity, translocase activation and protein secretion are coupled by one mechanism. We show here that a previously identified small molecule (HSI#6) binds SecA, modulates its intrinsic dynamics and allosterically activates the translocase in the absence of clients. By uncoupling translocase activation from preprotein binding, HSI#6 transformed the translocase into a promiscuous nanomachine that lost client selectivity and secreted unfolded pre- mature- and cytoplasmic- proteins with high efficiency in vivo or in vitro. To our knowledge, HSI#6 is the first activator of the Sec pathway and might offer unique opportunities for the discovery of new antibacterials. A small molecule that binds on SecA uncouples activation of the translocase from preprotein binding, transforming the Sec translocase into a promiscuous nanomachine that lost client selectivity and secretes any unfolded protein in vivo or in vitro.
|
|
Scooped by
mhryu@live.com
February 6, 4:37 PM
|
It is widely accepted that the use of siderophores, small molecules that bind and solubilize iron, emerged as a response to the dramatic reduction in bioavailability of this metal in aquatic environments caused by precipitation of iron oxides associated with the Great Oxidation Event (GOE). Here, we report a molecular clock analysis of the time of emergence of siderophore biosynthesis and utilization genes that challenges this view and argues for an emergence of these secondary metabolites that largely predates GOE. The emergence date of NIS siderophore synthases is found to predate by more than 1 Gy the emergence date of ferric siderophore reductases and esterases, which in turn also predate the GOE by approximately1Gy. This temporal gap is surprising given that these enzymes are essential for microorganisms to obtain iron from siderophores. This timing of events raises questions on the original ecological drivers for the emergence of siderophores. We offer an alternative hypothesis for the origin of siderophores which is their use in ferric mineral dissolution to avoid incrustation of neutrophilic iron oxidizers by metabolically generated ferric iron minerals. The observations and hypothesis reported here highlight the importance of environmental microbe-mineral interactions, beyond nutrient acquisition, as critical selective forces in early Earth, and call for a reassessment of the timing and drivers of siderophore evolution.
|
|
Scooped by
mhryu@live.com
February 6, 4:20 PM
|
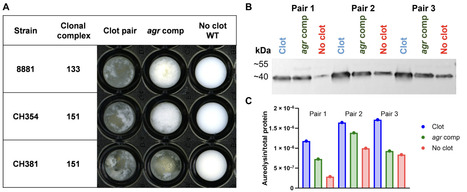
New pathogens typically arise from host jump events between species. Staphylococcus aureus is a multihost pathogen responsible for a global burden of human disease and a leading cause of intramammary infection in dairy cattle. Here, we demonstrate that following historical human-to-bovine host switch events, S. aureus has undergone adaptive metabolic remodeling in response to distinct nutrient availability in the dairy niche. In particular, we found that bovine S. aureus has evolved the capacity for protease-mediated degradation of casein, a protein abundant in bovine milk, to access nutrients for proliferation. This phenotype has evolved convergently in different S. aureus lineages via mutations in distinct gene loci driving overexpression of the protease aureolysin. Together, we have dissected a key host-adaptive trait, which facilitates the enzymatic release of nutrients from a substrate specific to the new host milieu. These findings highlight the remarkable evolutionary plasticity of a major bacterial pathogen underpinning its multihost species tropism.
|
|
Scooped by
mhryu@live.com
February 6, 3:57 PM
|
Evolutionary conservation has been considered a hallmark of essential basic functions in cells. Therefore, the study of evolutionarily conserved post-translational modifications (PTMs) can provide insight into their role in protein function. In this context, mass spectrometry can identify and quantify thousands of PTM sites. However, a major bottleneck lies in analyzing the large amounts of data collected by the mass spectrometer. Here we address the need for a protein sequence alignment tool for multiple PTMs across several species. We developed a tool named PTMOverlay that takes peptide identification output files and overlays PTM sites onto multiple protein sequence alignments. Examining 31 bacteria isolates, we combined their protein sequences with select PTM types, including acetylation, phosphorylation, monomethylation, dimethylation, and trimethylation. The tool revealed a variety of conserved modification sites on the bacterial central carbon metabolism. Further structural analysis revealed possible interactions between methylated arginine and lysine residues with phosphothreonine/serine sites on the homodimer interface of enolase. Overall, this tool can parse large amounts of mass spectrometry data and allows for more informed and efficient selection of sites for future studies of protein function.
|
|
Scooped by
mhryu@live.com
February 6, 3:40 PM
|
Understanding how evolutionary constraints shape protein sequences is fundamental to deciphering the molecular mechanisms underlying protein stability and function, which has broad implications in protein engineering and therapeutics development. Recent advances in protein language models (pLMs) have enabled accurate prediction of mutation effects through evolutionary information, effectively capturing the selective pressure that governs protein sequence variation. A critical challenge, however, remains in disentangling the intertwined mutation effects on protein stability and function, as evolutionary signals conflate both stability-driven and function-driven pressures, obscuring the mechanistic basis of mutation effects and limiting their utility for rational protein engineering. In this work, we introduce DETANGO, a novel deep learning framework that explicitly deconvolves the mutation effects on protein functions by removing components attributable to stability perturbations from the pLM-predicted mutation effects. Guided by computational or experimental stability measurements, DETANGO estimates a functional plausibility score for each single-point mutation that is the component of the mutation effect not accounted for by changes in stability. Single-point mutations with low functional plausibilities are predicted to be stable-but-inactive (SBI) variants, whose compromised activities are caused by direct perturbations on functional mechanisms rather than structural stability. Residues enriched for such variants are inferred to be functionally critical, as indicated by the strong evolutionary pressures to maintain protein function. Through extensive benchmarking experiments, we show that DETANGO accurately identifies SBI variants and pinpoints functionally important residues across contexts, including ligand binding, catalysis, and allostery. Moreover, extending DETANGO from individual proteins to homologous protein families reveals shared and distinctive functional patterns across protein families. Collectively, these results establish DETANGO as a biologically grounded framework for disentangling evolutionary constraints on protein stability and function, advancing mechanistic understanding of protein function, and informing rational protein engineering.
|
|
Scooped by
mhryu@live.com
February 6, 3:30 PM
|
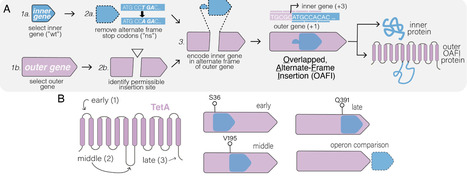
Overlapping genes—wherein two different proteins are translated from alternative reading frames of the same DNA sequence—provide a means to stabilize an engineered gene by directly linking its evolutionary fate with that of an overlapping gene. However, creating overlapping gene pairs is challenging, as it requires redesigning both protein products to accommodate overlap constraints. Here, we present a new “overlapping, alternate-frame insertion” (OAFI) method for creating synthetic overlapping genes by inserting an “inner” gene, encoded in an alternate frame, into a flexible region of an “outer” gene. Using OAFI, we create new overlapping gene pairs of genetic reporters and bacterial toxins within an antibiotic resistance gene. We show that both the inner and outer genes retain function despite redesign, with translation of the inner gene influenced by its overlap position in the outer gene. Importantly, we show that, despite these inner gene sequences not contributing to outer gene function, selection for the outer gene alters the permitted inactivating mutations in the inner gene, and that overlapping toxins can restrict horizontal gene transfer of the antibiotic resistance gene. Overall, OAFI offers a versatile tool for synthetic biology, expanding the applications of overlapping genes in gene stabilization and biocontainment.
|
|
Scooped by
mhryu@live.com
February 6, 2:52 PM
|
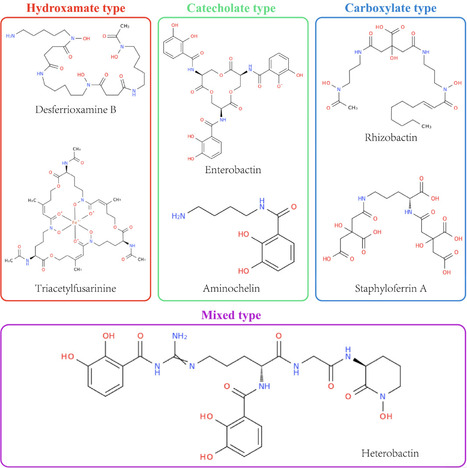
Iron is an essential micronutrient for nearly all microorganisms, yet its bioavailability is severely limited in most environments. To overcome this restriction, microorganisms produce siderophores, high-affinity iron-chelating molecules that play pivotal roles in microbial iron homeostasis, interspecies competition, and host–pathogen interactions. Despite extensive research, current understanding of siderophore biosynthetic and regulatory diversity remains largely limited to specific models, with comprehensive cross-taxonomic frameworks only beginning to emerge. This review systematically integrates recent advances in the genetic and biochemical foundations of microbial siderophore production, focusing on the two major biosynthetic pathways: nonribosomal peptide synthetase (NRPS)-dependent and NRPS-independent synthetase (NIS). We further elaborate on the diverse transport systems in Gram-negative and Gram-positive bacteria, as well as fungi, alongside the iron-responsive regulators (e.g., Fur) and gene clusters that coordinate iron uptake and utilization. Beyond physiological mechanisms, we discuss how these insights inform emerging applications of siderophores across multiple fields: in medicine, enabling “Trojan horse” antimicrobial strategies; in agriculture, enhancing plant iron uptake and serving as biocontrol agents; in environmental remediation, facilitating heavy-metal detoxification; and in biosensing, acting as selective probes for metals and pathogens. By bridging fundamental mechanisms with practical applications, this review aims to provide an integrative perspective for future exploration of microbial iron homeostasis and its biotechnological potential.
|
|
Scooped by
mhryu@live.com
February 6, 2:30 PM
|
The growing global demand for food is limited by low seed germination rates, a key constraint in crop production. Priestia megaterium W101, a plant growth-promoting rhizobacterium with high indole-3-acetic acid (IAA) yield, was found to significantly promote the germination of wheat seeds and the growth of the root system. For the first time, we characterized in Priestia megaterium a complete indole-3-pyruvic acid (IPyA) pathway, encoded by ipdC, patB, feaB, and gene1566, together with a yedL-encoded alternative pathway, through integrated multiomics analyses and CRISPR/Cas9-mediated heterologous expression. In addition, we reconstructed the complete IPyA pathway in Bacillus subtilis 168, which increased IAA production by approximately 98% compared to that in the wild-type strain. Overall, this study elucidates the IAA biosynthetic network in W101 and highlights its potential as a core strain for sustainable microbial inoculant development in green agriculture.
|
|
Scooped by
mhryu@live.com
February 6, 1:53 PM
|
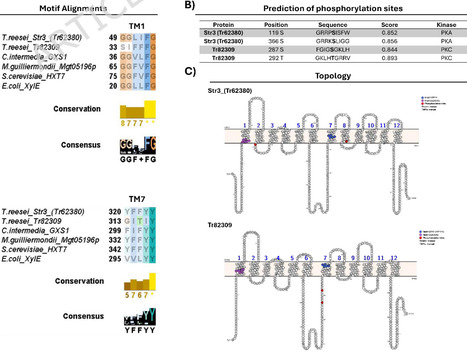
The engineering of Saccharomyces cerevisiae for the use of xylose is fundamental to improving fermentation performance in the production of second-generation ethanol (2G) via pentose fermentation. For this, one of the main strategies involves expressing heterologous xylose transporters to ensure efficient uptake of this sugar. However, due to the intrinsic non-specificity of sugar transporters, competition occurs between sugars (e.g., xylose and glucose), leading to reduced pentose transport efficiency and lower ethanol productivity. This study aimed to develop and characterize sugar transporters with lower affinity for glucose, while maintaining the ability to transport xylose, through genetic improvement of Trichoderma reesei transporters for heterologous expression in S. cerevisiae. To this end, alignments were made to find motifs described as important for xylose transport, and phosphorylation sites were predicted to achieve the objective. Based on these predictions, the transporters were modeled and docked with glucose and xylose. The transporters with the desired phenotype were expressed in S. cerevisiae strains for characterization. Drop assays and aerobic fermentation trials were performed to confirm the predicted profile. In silico analysis shows that two mutations in Str3 (Tr62380) exhibited a promising phenotype. For Tr82309, which has not yet been characterized, it was decided to proceed with characterizing the wild transporter. Drop assays revealed qualitative differences in growth phenotypes among the tested transporters. Docking was used strictly as a qualitative, hypothesis-generating tool to prioritize mutations for experimental testing and was not intended to predict transport kinetics or substrate affinity. The mutants of Str3 (Tr62380) did indeed lose their natural affinity for hexoses. Additionally, Tr82309 showed limited activity in liquid assays but supported growth at higher xylose concentrations in solid medium, suggesting condition-dependent functionality rather than confirmed substrate specificity. In the aerobic fermentation assay, only Str3 (Tr62380)_WT exhibited high efficiency in the uptake of sugars from the medium; mutations inserted in Str3 (Tr62380) reduced the ability to transport sugars, primarily glucose. Phosphorylation mimetics provided in vivo evidence that post-translational modification can influence the substrate utilization profile of sugar transporters. Docking served as a qualitative screening step to guide the selection of mutations for experimental validation. We also present phosphorylation sites as a new target for engineering studies of sugar transporters. However, experimental validation is indispensable. In summary, our findings provide a rational framework for the engineering of glucose-insensitive xylose transporters, enabling more efficient co-fermentation in biorefineries.
|
|
Scooped by
mhryu@live.com
February 6, 1:25 PM
|
How long do fungal hyphae persist in the environment? And how does this differ between groups and species of fungi? Despite growing knowledge of fungal contributions to decomposition and soil carbon cycles, surprisingly little is known about the turnover of mycelia: What happens to fungal hyphae over time? And how this impacts different fungi's contribution to carbon sequestration? In this study, we compared microscale persistence of fungal hyphae using microfluidic chip technology and visual quantification of hyphal degradation and turnover across six different wood-decay Basidiomycete species. Measured traits included hyphal extension, coverage, turnover rates, and changes in hyphal morphology over time when supplied with two carbon sources of differing recalcitrance. Species clustered into two groups: one with a frugal nutrient strategy (high turnover capacity, active persistence of cytoplasmic hyphae) and one with a wasteful strategy (low turnover of hyphae and large remnants of skeletonized hyphae). Differences matched the ephemeral or long-lasting nature of their fruiting bodies and the substrates they inhabit. Carbon type also influenced hyphal persistence over time. Our results suggest that hyphal turnover has a genetic basis linked to species ecology yet is also shaped by environmental factors such as carbon availability, highlighting the dynamic nature of fungal mycelia.
|
|
Scooped by
mhryu@live.com
February 6, 1:16 PM
|
Plant-biotic interactions are driven by the exchange of molecules. Small peptide hormones like CLAVATA3/EMBRYO SURROUNDING REGION (CLE) peptides play central regulatory roles in these interactions. CLEs determine the extent of symbiotic interaction to balance costs and benefits for the host. In parasitic interactions, CLEs regulate the formation of feeding sites by plant pathogenic nematodes and promote the formation of haustoria in parasitic plants. By reviewing recent findings on CLE functions, their receptors, and responses across different biotic interactions, we provide insights into the increasingly complex roles of CLEs in plant development and nutrient signaling.
|
|
Scooped by
mhryu@live.com
February 6, 9:59 AM
|
Recombinant E. coli with nitrogen-fixation activity has been constructed using a simple approach of introducing a minimal gene set of nitrogen fixation genes along with its upstream region from Paenibacillus species; however, limitations may exist in nif expression. To understand the limitations of heterologous mass production of nitrogenase in E. coli, we evaluated this approach using various nif operons cloned from Paenibacillus species. Initial attempts revealed that, despite the high nitrogenase activity observed in the parental strains, the corresponding nif operons did not consistently function in E. coli. Further analysis revealed that certain upstream regions of the nif operons function as constitutive σ70-dependent promoters in E. coli, and the host acquired nitrogenase activity only when the upstream region acted as a constitutive promoter. Notably, recombinant E. coli achieved high nitrogenase activity by linking a functional upstream region to the nif operon from a highly active parental strain. Additional improvements in nitrogenase activity were achieved by combining promoter replacement with synthetic biology approaches to enhance electron transfer from glucose metabolism to nitrogenase and metal cluster assembly for protein maturation. These findings provide new insights into heterologous nitrogenase production in E. coli for sustainable nitrogen fixation.
|
|
|
Scooped by
mhryu@live.com
February 6, 4:45 PM
|
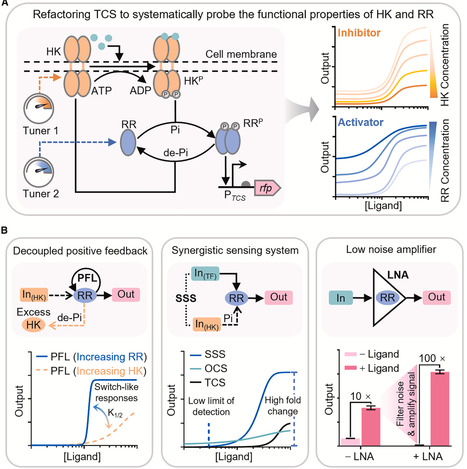
Two-component systems (TCS) are ubiquitous multi-step signal sensing systems in prokaryotes and are promising platforms for building cellular sensors. However, their programmability remains underexplored, limiting broader applications in synthetic biology. Here, we refactor TCSs to systematically elucidate the functional properties of response regulator (RR) and histidine kinase (HK) as the concentration-dependent activator and inhibitor for TCS sensor output, respectively. By decoupling HK expression from native feedback circuitry, we engineer ultrasensitive TCS sensors with tunable detection thresholds. By leveraging RR as a transducer, we couple one-component system (OCS) and TCS to create a synergistic sensing system (SSS) characterized by both a low detection limit and a high dynamic range. We further show that RR alone serves as a biological-low noise amplifier (LNA), substantially upgrading performance of diverse genetically encoded biosensors. Our study demonstrates TCS’s high plasticity and programmability for customizing gene expression regulation in synthetic circuits, providing modular toolkits for biosensor optimization.
|
|
Scooped by
mhryu@live.com
February 6, 4:39 PM
|
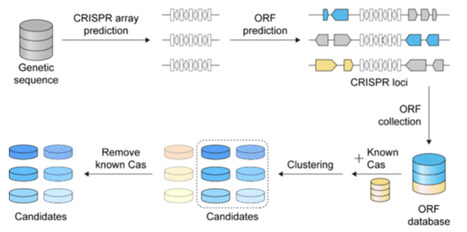
Efficient delivery remains a major challenge for therapeutic genome editing because many widely used CRISPR nucleases are large and leave limited space for regulatory elements or additional payloads in a single adeno-associated virus (AAV) vector. Miniature Cas12 nucleases are particularly appealing, as their reduced size alleviates packaging constraints while preserving RNA-guided DNA cleavage. Here, we outline a workflow that links large-scale sequence mining with phylogenetic and structural filtering, followed by PAM profiling, in vitro cleavage, bacterial genome interference, and genome-editing assays in human cells to confirm activity. This protocol is intended to distill broad sequence collections into a small set of compact Cas12 nucleases with demonstrated functions that can serve as starting points for further engineering in delivery-limited settings.
|
|
Scooped by
mhryu@live.com
February 6, 4:34 PM
|
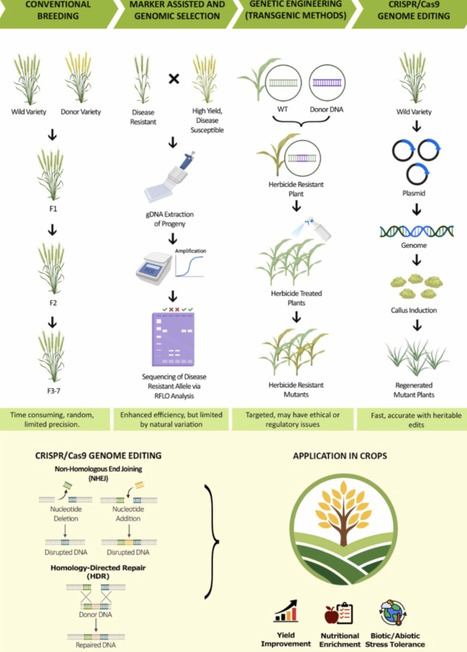
Global food security is escalating by population growth, climate change and depletion of basic resources, and explicitly demands the implementation of cutting-edge approaches to improve crop yield, resilience, and nutritional quality. CRISPR/Cas9 technology has transformed modern agriculture by introducing accurate and inherently stable modifications in different plants. This review highlights the latest advancements in the application of CRISPR/Cas9 technology for crop improvement and explores its potential in mitigating global food security. These advancements include the use of base and prime editing to accurately alter metabolic pathways for nutritional enhancements, along with designing Cas variants with limited dependency on PAM, to facilitate editing in complex genome crops like wheat. Moreover, the integration of artificial intelligence-driven target prediction and speed breeding has significantly improved varietal development by shortening breeding period and increasing resilience to various biotic and abiotic stresses. Case studies in cereal (Rice, wheat, maize, and sorghum) and horticultural crops provide evidence of CRISPR’s major contribution towards limiting food security, improving nutritional value, and mitigating postharvest waste. This section also addresses the dynamic regulatory developments in different areas, associated ethical reflections, and approaches to foster fair accessibility stressing the transparent governance and public participation in the implementation of this technique.
|
|
Scooped by
mhryu@live.com
February 6, 4:06 PM
|
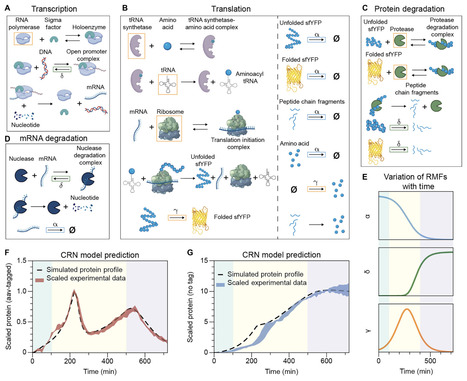
Synthetic gene circuits often behave unpredictably in batch cultures, where shifting physiological states are rarely accounted for in conventional models. Here, we find that degradation-tagged protein reporters could exhibit transient oscillatory expression, which standard single-scale models do not capture. We resolve this discrepancy by developing Gene Expression Across Growth Stages (GEAGS), a dual-scale modeling framework that explicitly couples intracellular gene expression to logistic population growth. Using a chemical reaction network model with growth phase–dependent rate-modifying functions, GEAGS accurately reproduces the observed transient oscillations and identifies amino acid recycling and growth-phase transition as key drivers. We reduce the model to an effective form for practical use and demonstrate its adaptability by applying it to layered feedback circuits, resolving long-standing mismatches between model predictions and measured dynamics. These results establish GEAGS as a generalizable platform for predicting emergent behaviors in synthetic gene circuits and underscore the importance of multiscale modeling for robust circuit design in dynamic environments.
|
|
Scooped by
mhryu@live.com
February 6, 3:52 PM
|
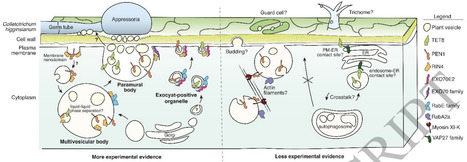
Extracellular vesicles (EVs) produced by Arabidopsis (A. thaliana) plants are highly heterogeneous in protein content. To understand the origins of plant EV heterogeneity, we used an unbiased proximity labeling approach to identify proteins and pathways involved in the secretion of distinct EV subpopulations. Proximity labeling, co-immunoprecipitation, and fluorescence microscopy in Nicotiana benthamiana all indicated a general role in EV secretion for EXO70 proteins (a subunit of the exocyst complex) and the immune-related protein RPM1-INTERACTING PROTEIN4 (RIN4). To confirm these hypotheses, we assessed the impact of mutations in various EXO70 family genes and in RIN4 on EV release, as well as mutations in additional genes known to regulate endomembrane trafficking and secretion. Mutation of EXO70E1, EXO70E2 or STOMATAL CYTOKINESIS DEFECTIVE1 (SCD1; a GTP-exchange factor for RabE GTPases) reduced secretion of EVs marked by TETRASPANIN8 (TET8), PENETRATION1 (PEN1), and PATELLIN1 (PATL1), indicating that these proteins are generally required for EV secretion. In contrast, mutation of RIN4 reduced levels of TET8+ and PEN1+ EVs, but not PATL+ EVs. Mutation of the small GTPase gene RABA2a specifically affected PEN1+ EV secretion, while mutations in AUTOPHAGY PROTEIN5 (ATG5) and VAMP-ASSOCIATED PROTEIN27 (VAP27) specifically affected TET8+ EV secretion. Lastly, we found that exo70 family mutants are more susceptible to infection with the fungal pathogen Colletotrichum higginsianum, underlining the importance of secretion for plant immunity. Together, our results unravel some of the complex mechanisms that give rise to EV subpopulations in plants.
|
|
Scooped by
mhryu@live.com
February 6, 3:34 PM
|
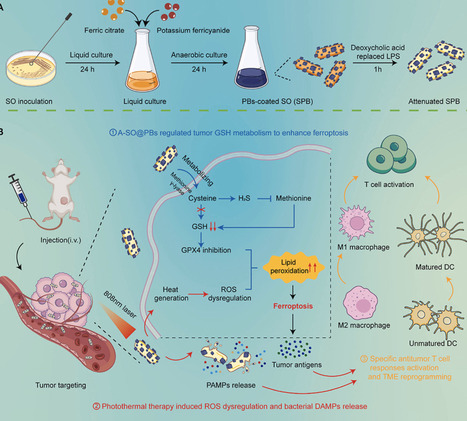
Bacterial combination therapy offers immense promise for treating aggressive "cold" solid tumors, such as triple-negative breast cancer (TNBC). However, the clinical translatability of traditional genetic engineering is often hampered by operational complexity and genetic instability. Here, we developed A-SPB—a non-genetically engineered, multi-functional living bioreactor based on Shewanella oneidensis MR-1 (SO), surface-modified with Prussian blue (PB) nanoparticles and attenuated via deoxycholic acid (DA) treatment. This platform serves as an in situ metabolic factory that "hijacks" the sulfur-metabolism within the hypoxic tumor microenvironment. By actively depleting cysteine, A-SPB not only starves the glutathione (GSH) synthesis pathway but also generates hydrogen sulfide (H2S) to inhibit the transsulfuration bypass, leading to a dual-pronged collapse of the GSH/GPX4 antioxidant axis and triggering robust tumor ferroptosis. This metabolic sensitization is further amplified by the PB-mediated photothermal therapy (PTT), which generates localized hyperthermia and excessive reactive oxygen species (ROS). Notably, the PTT serves as a dual-functional "bio-switch": it promotes acute tumor ablation while simultaneously triggering bacterial self-lysis to ensure biosafety. This programmed lysis releases a synergistic cocktail of bacterial PAMPs and tumor-derived DAMPs, which effectively remodels the immunosuppressive TME and initiates a potent systemic antitumor immune response. By integrating metabolic reprogramming, sensitized ferroptosis, and on-demand immune activation through simple surface engineering, this study provides a highly translatable and safe paradigm for the next generation of living bacterial therapeutics.
|
|
Scooped by
mhryu@live.com
February 6, 3:01 PM
|
Microbes inhabiting soils experience periodic water deprivation. The effects of desiccation on DNA, protein, and membrane integrity are well-described. However, the effects of drying and rehydration on the composition of cellular RNA and metabolites are still poorly understood. Here, we describe how slow drying and rehydration with water vapor influence the composition of RNAs and metabolites in a soil Arthrobacter. While drying reduced cultivability relative to hydrated controls, water vapor rehydration fully restored it. Ribosomal RNA proportions remained constant throughout all treatments, and mRNA profiles showed stable composition during desiccation—changing only during transitions into and out of desiccation-induced dormancy. Six transcriptional modules displayed distinct expression patterns in desiccated-rehydrated samples relative to hydrated controls, including desiccation-rehydration responsive and rehydration-specific profiles. Targeted intracellular metabolomics revealed similarly static profiles during desiccation, with a cluster of ribonucleosides and nucleobases increasing in response to desiccation and returning to baseline levels upon rehydration with water vapor. These findings demonstrate that both mRNA and metabolite profiles remain essentially frozen in desiccated Arthrobacter, with dynamic changes occurring only during state transitions. These results have important implications for environments with frequent drying cycles where stable mRNA in dormant cells combined with intracellular RNA recycling may obscure interpretations of RNA-based environmental analyses that use RNA as a marker of microbial activity. Our results suggest that RNA-based activity assessments in periodically dry environments require careful consideration of dormancy-associated molecular preservation.
|
|
Scooped by
mhryu@live.com
February 6, 2:30 PM
|
Cassava production in sub-Saharan Africa is severely impacted by diseases. Most pathogens require interaction with host susceptibility factors to complete their life cycles and cause disease. Targeted DNA methylation is an epigenetic strategy to alter gene expression in plants, and we previously reported that a zinc-finger fused to DMS3 could establish methylation at the promoter of MeSWEET10a, a bacterial susceptibility gene, and this resulted in decreased disease. Here, we attempt a similar strategy for cassava brown streak disease. This disease is caused by the ipomoviruses CBSV and UCBSV. These viruses belong to the family Potyviridae, which has been shown extensively to require host eIF4E-family proteins to infect plants and cause disease. We previously found that cassava plants with simultaneous knockout mutations in two eIF4E genes, nCBP-1 and nCBP-2, resulted in decreased susceptibility to CBSD. Here, we report successful simultaneous targeting of both promoters with methylation using a dCas9-DRMcd-SunTag system. However, in contrast to our previous work with MeSWEET10a, controls indicate that CRISPR interference is occurring in these lines and is sufficient for the reduction of gene expression. Future research will use genetic crosses to segregate away the DNA methylation reagents and, if DNA methylation proves heritable, assess whether methylation alone is sufficient to increase resistance to CBSD.
|
|
Scooped by
mhryu@live.com
February 6, 2:00 PM
|
This work constructed a recombinant lactic acid bacterium secreting β-galactosidase for GOS formation during milk fermentation. First, GalINF, a β-galactosidase derived from infant feces, was characterized to effectively produce GOS in milk, reaching a content of 10.03 g/L. To export GalINF in Lactococcus lactis, six signal peptide candidates were employed, resulting in extracellular activities as low as 52.83–85.65 U/L. Then, GalINF (114.6 kDa) was split into two complementary modules, M1-P723 and A724-I1023, which could be independently secreted and actively reconstituted with the help of the protein scaffold SpyCatcher/SpyTag. The resultant Lc. lactis B1RG exhibited an extracellular β-galactosidase activity of 544.22 U/L. Fermentation of pasteurized milk with Lc. lactis B1RG and the traditional yogurt starters reduced lactose to 19.67 g/L and yielded 7.17 g/L GOS. This work established an effective strategy to export large-sized proteins extracellularly and demonstrated the applicability of LAB secreting β-galactosidase for GOS-enriched fermented dairy products.
|
|
Scooped by
mhryu@live.com
February 6, 1:35 PM
|
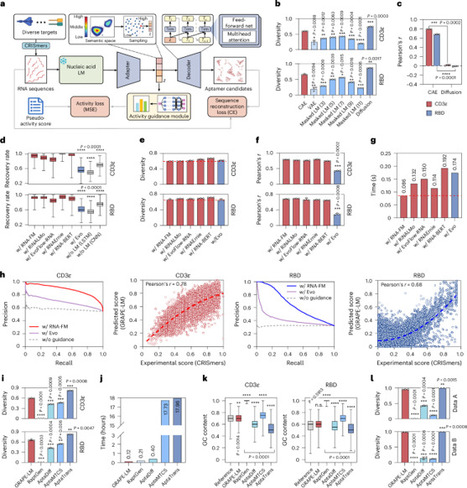
The directed evolution of biomolecules is an iterative process. Although advancements in language models have expedited protein evolution, effectively evolving RNA remains a challenge. RNA aptamers, selected for their binding properties, provide an ideal system to address this challenge, yet traditional aptamer discovery still relies on labor-intensive, multi-round screening. Here we introduce GRAPE-LM (generator of RNA aptamers powered by activity-guided evolution and language model), a generative artificial intelligence framework designed for the one-round evolution of RNA aptamers. GRAPE-LM integrates a transformer-based conditional autoencoder with nucleic acid language models and is guided by CRISPR−Cas-based aptamer screening data derived from intracellular environments. We validate GRAPE-LM on three disparate targets: the human T cell receptor CD3ε, the receptor-binding domain of the SARS-CoV-2 spike protein and the human oncogenic transcription factor c-Myc (an intracellular disordered protein). GRAPE-LM, informed with only a single round of CRISPR−Cas-based screening, successfully obtains RNA aptamers that outperform those driven from multiple rounds of human selection and optimization. Combining generative AI and one round of wet lab evolution generates high-affinity RNA aptamers.
|
|
Scooped by
mhryu@live.com
February 6, 1:21 PM
|
Crop protection against soilborne pathogens such as root-knot nematodes (Meloidogyne spp.) has been a losing battle on too many occasions. Often going undetected until they cause major crop losses, these organisms have been conventionally managed by field application of synthetic nematicides, with high environmental cost. Agroecologically sound practices for nematode control, including soil organic matter management, crop diversification and intercropping, are being developed and tested, but the underlying mechanisms behind successful control have remained elusive. In an article, Hama et al. (2025; doi:10.1111/nph.70789) have uncovered a form of plant resistance that may be responsible for the complex and often unanticipated effects in the field: plants can transfer defence compounds between them, causing significant metabolome changes and, ultimately, inducing root-knot nematode resistance in otherwise susceptible neighboring plants. benzoxazinoids (BXs) exuded by rye (Secale cereale) roots into the soil can be taken up, metabolised and translocated by roots of clover (Trifolium repens)
|
|
Scooped by
mhryu@live.com
February 6, 1:09 PM
|
The extent to which microbial processes control soil organic carbon (SOC) dynamics remains uncertain. Carbon use efficiency (CUE), that is, the fraction of assimilated carbon allocated to growth, has been used as a key parameter but its relationship with SOC reflects carbon partitioning rather than the absolute magnitude of microbial fluxes. The microbial growth rate could provide a more mechanistic link to SOC accumulation because it quantifies biomass production and reflects necromass formation. Here we combine a global ¹⁸O–H2O dataset (n = 268 paired observations) with outputs from four land surface models to test whether growth rate predicts SOC more strongly than CUE. In the incubation experiments, growth rates are more closely associated with SOC than CUE, although soil properties and climate explain equal or greater variance. Models reproduce the stronger role of growth rate over CUE but tend to underestimate the abiotic controls. The models also emphasize CUE as the main predictor of the SOC-to-net primary production ratio, in contrast to observations, which indicates the soil’s capacity to retain plant carbon inputs. Together, these findings identify the microbial growth rate as a diagnostic that can help bridge models with empirical data and guide a more balanced representation of microbial and mineral controls in SOC projections. Microbial carbon use efficiency is a strong predictor of soil organic carbon stocks. Here the authors reveal that the microbial growth rate is a more reliable and informative predictor, and that modelling approaches tend to overemphasize the role of biotic over abiotic controls compared to empirical data.
|