 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 10:45 PM
|
Structural variations (SVs), including inversions, translocations, duplications, and large insertions or deletions, are key drivers of genome evolution and phenotypic diversity. With the increasing number of high-quality, chromosome-scale genome assemblies, the ability to detect and interpret SVs has become a crucial aspect of modern genomics. While SV detection has advanced, most visualization methods produce static plots that fall short when researchers, particularly in comparative genomics, need to interactively explore large datasets, zoom into specific genomic regions, or dynamically filter structural events in real time. To address this gap, we introduce SynFlow, a lightweight, web-based interactive application specifically designed for exploring and visualizing structural variations identified by SyRI. We demonstrate that SynFlow can reproduce complex static synteny plots published in literature, but transforms them into dynamic, shareable visualizations that support real-time filtering, reordering, and deep exploration of specific SVs, including translocations. SynFlow is available as a web server and offers multiple entry points: browsing precomputed datasets (e.g., banana and grapevine genomes), uploading user-provided SyRI outputs, or running an integrated workflow to produce and visualize SVs on the fly.
|
|
Scooped by
mhryu@live.com
Today, 3:02 PM
|
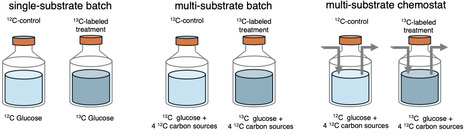
Quantitative stable isotope probing (qSIP) allows researchers to calculate taxon-specific carbon incorporation from sequencing of natural microbial communities, which can be used as a proxy for metabolic activity rates and subsequently as an input for biogeochemical modeling. While qSIP is widely utilized in soils to investigate the identity and metabolic activity of largely unculturable microbes, the application of qSIP in marine and aquatic ecosystems is more recent. Here, we investigated how bioreactor type (batch vs. chemostat) and carbon substrate complexity (single vs. multiple substrates) affect the incorporation of 13C-labeled glucose into rRNA after 24 hours using excess atomic fraction (EAF) as a proxy for metabolic activity rate. We found that the growth dynamics and community composition of the 13C-incorporating bacteria differed significantly for each treatment. EAF was positively correlated with both 16S gene copy number and a genomic index of copiotrophy in both batch treatments, but not in the chemostat, suggesting that chemostats dampen the competitive advantage of fast-growing copiotrophic taxa. Our results demonstrate that both substrate complexity and experimental regime influence qSIP-derived metabolic activity estimates and provide guidance for future applications of qSIP in aquatic environments.
|
|
Scooped by
mhryu@live.com
Today, 2:54 PM
|
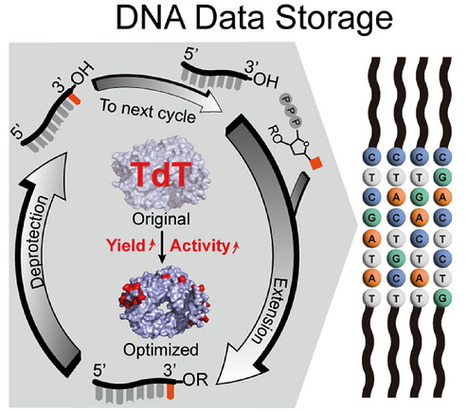
DNA, with its exceptional information capacity and chemical stability, represents a promising material for next-generation data storage to meet the exponential growth of global digital information demands. Enzymatic DNA synthesis provides a sustainable route to DNA production. However, the practical scalability of the underlying polymerization chemistry has been fundamentally constrained by the low catalytic efficiency and aggregation-induced inactivation of terminal deoxynucleotidyl transferase (TdT) that is responsible for nucleotide polymerization. Here, we report a structure-guided enzyme design framework that overcomes these intrinsic limitations by decoupling solubility and catalytic performance in a processive polymerase. Computational redesign of aggregation-prone regions markedly enhances soluble expression, while targeted active-site engineering improves catalytic efficiency toward 3′-ONH2-dNTPs used in enzymatic DNA synthesis. The resulting TdT variant HL2-LKI achieves 3.4 g L−1 soluble expression in a 5 L fermenter without fusion tags and exhibits high polymerization efficiency (99.9%) and DNA writing fidelity (98.9%). This redesign reduces enzyme production costs to approximately $0.7 g−1, nearly seven orders of magnitude lower than the catalog price of commercially available TdT. This work establishes a generalizable strategy for transforming aggregation-limited enzymatic polymerization reactions into scalable and low-cost molecular manufacturing processes, thereby advancing the practical implementation of DNA as an information material.
|
|
Scooped by
mhryu@live.com
Today, 2:49 PM
|
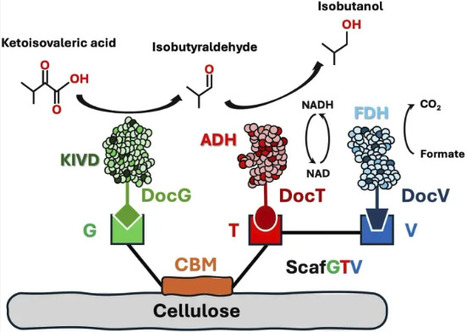
Increasing environmental and economic pressures associated with global fossil fuel demand necessitate a shift toward sustainable fuel production. Production of second-generation biofuels, such as isobutanol, presents a promising opportunity; however, product toxicity limits in vivo production, motivating the development of optimized in vitro systems. As prior efforts have focused on enzyme engineering to improve titer, these systems remain constrained by diffusion-limited mass transfer. Here, we introduce an engineered, ordered cellulosome-based immobilization system that facilitates enhanced enzymatic productivity. Using keto-acid decarboxylase, alcohol dehydrogenase, and formate dehydrogenase as a model for the final steps of the isobutanol pathway, the system achieved a preliminary isobutanol titer of 5.92 g/L, a 78.4% yield, and an enzymatic productivity of 0.34 mL-1 h-1, representing marked improvement over previous cell-free approaches. This proof-of-concept approach introduces the importance of targeted immobilization alongside enzyme optimization and demonstrates ordered scaffoldin-mediated immobilization as a versatile platform approach for future in vitro biofuel and biochemical production.
|
|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
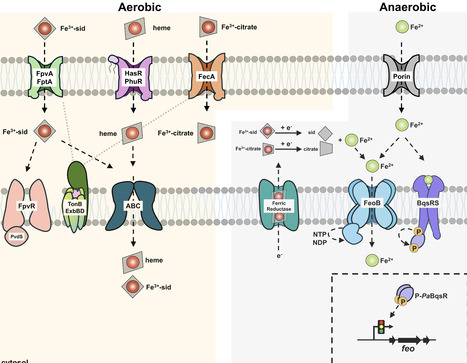
Pseudomonas aeruginosa is a ubiquitous, Gram-negative bacterium that forms biofilms and is responsible for antibiotic-resistant hospital-acquired infections in humans. The P. aeruginosa BqsRS two-component system regulates biofilm formation and dispersal by sensing extracytoplasmic Fe2+, but the mechanistic details of this process are poorly understood. In this work, we report the crystal and solution structures of the PaBqsR response regulator receiver domain, comprising a (βα)5 response regulator assembly, and the DNA-binding domain, comprising a helix-turn-helix motif. Consistent with its cognate stimulus being Fe2+, we show that BqsR binds directly to the promoter region of the feo operon that encodes the bacterial Fe2+ transport system FeoABC. Corroborating these in vitro results, transcriptional studies show that BqsR is a global regulator controlling many important genes in PAO1, including the feo operon. Intriguingly, promoter-based assays reveal that BqsR is a dynamic regulator that responds to bioavailable Fe2+, likely through the ability of BqsR to bind Fe2+ directly via a His-rich motif, independent of the BqsS membrane His kinase. To our knowledge, this mode of regulation has not been reported previously among OmpR-like response regulators but represents an important level of control over Fe2+ acquisition in P. aeruginosa that could be an attractive therapeutic target to treat hospital-acquired infections. The BqsRS two-component system regulates biofilms in the infectious pathogen Pseudomonas aeruginosa. Here, the authors show that the global response regulator BqsR controls Fe2+ acquisition in PAO1 through a previously unrecognized regulatory mechanism.
|
|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
Artificial intelligence (AI) is transforming scientific research, including proteomics. In this Perspective, we highlight key mass spectrometry (MS)-based proteomics areas where AI is driving innovation, ranging from protein identification to building AI virtual cells. These include improving peptide and protein identification and quantification; characterizing protein-protein interactions and protein complexes; advancing spatial and perturbation proteomics; integrating multi-omics data; and, ultimately, enabling AI virtual cells. Finally, we call for global collaboration among data producers, data consumers and other stakeholders to establish an AI-friendly ecosystem for MS-based proteomics, laying the foundation for transformative advancements in proteomics driven by AI. This Perspective highlights key research areas within mass spectrometry-based proteomics where AI is poised to drive significant advances.
|
|
Scooped by
mhryu@live.com
Today, 1:19 AM
|
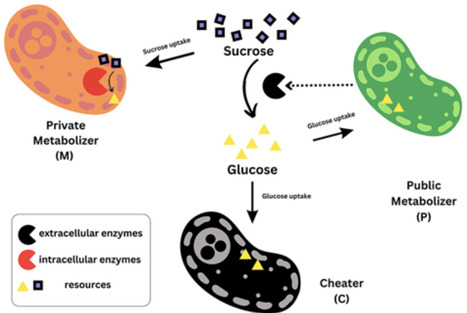
This study presents a mathematical framework for investigating the dynamics of coexistence and competition among heterotrophic microbes across different time scales. Focusing on metabolic interactions, we examine how three strategies, public metabolizing, private metabolizing and cheating, shape population behavior. The framework integrates generalized Lotka–Volterra dynamics with evolutionary game theory to capture the effects of resource exchange, particularly glucose made available by public metabolizers and sucrose as a shared substrate driving population growth. Game-theoretic pay-offs encode ecological costs and benefits, enabling analysis of frequency-dependent interactions among strategies. To capture evolutionary realism, we implement laboratory-inspired simulations in which strategies can switch between generations, mimicking mutation or phenotypic plasticity in microbial populations. These eco-evolutionary dynamics reveal conditions under which all three strategies coexist at interior equilibria and show how variation in growth advantages and, illustratively, phenotype-switching perturbations produce evolutionary shifts. Numerical analysis identifies ecological thresholds and fitness asymmetries that determine system robustness, long-term coexistence, and the persistence of a synthetic, cross-kingdom system linked by nutrient exchange. Together, these insights provide general principles for microbial coexistence and offer design guidelines for ecosystem engineering, biotechnological applications and the construction of stable synthetic communities under ecological and evolutionary constraints.
|
|
Scooped by
mhryu@live.com
Today, 1:02 AM
|
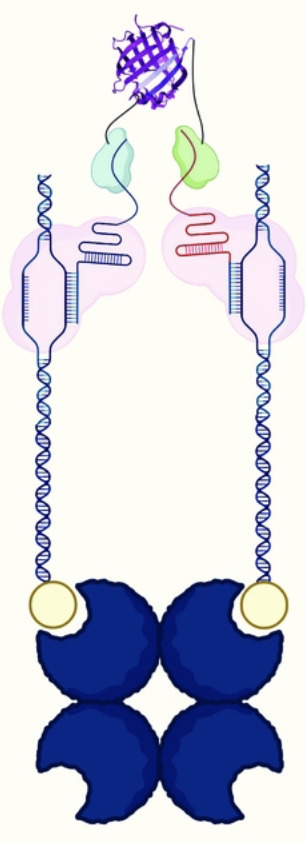
The dCas9 system has rapidly been developed into many tools to explore different aspects of the human genome; the high binding specificity, coupled with the inactive nuclease enzyme, allows for precise recruitment of molecules to specific sequences of DNA. We sought to exploit these capabilities to create a tool for assessing the real-time proximity of two DNA sequences within a biological system. By incorporating aptamers into gRNAs, dCas9 molecules can be used to recruit the β9 or β10 strands of split-NanoLuc® to specific DNA sequences and quantify the proximity of those sequences based on their ability to complex with the luciferase fragment (Δ11S) and produce luminescence. While many tools exist to detect a single DNA sequence, this system is uniquely capable of assessing how two DNA sequences interact with each other. As expected, we found that the interaction of two dCas9 molecules was affected by their linear distance from each other on dsDNA. Surprisingly, we found that their interaction was also strongly influenced by rotational orientation, even for sequences that are close together in linear space. This finding indicates that dCas9 rotational alignment is an important consideration for designing dCas9 systems that target multiple DNA sequences simultaneously. Beyond the findings presented herein, we believe this DNA proximity detection tool has the potential to be adapted for applications involving the proximity and orientation of two DNA sequences.
|
|
Scooped by
mhryu@live.com
Today, 12:56 AM
|
Methane (CH4) from ruminants is a major source of agricultural greenhouse gas and represents a loss of dietary energy. 3-nitrooxypropanol (3-NOP) is a known methanogenesis inhibitor, but its hydrophilic nature may limit the cellular accessibility to methanogens. Here, we systematically evaluated 1,3-propanediol dinitrate (1,3-PDN), a more hydrophobic derivative of 3-NOP, for its anti-methanogenic potential and underlying mode of action using ruminal fermentation, pure-culture assays, multi-omics analyses, and molecular docking. Intracellular accumulation assays indicated greater cellular accumulation of 1,3-PDN than 3-NOP in rumen-derived methanogen Methanobrevibacter olleyae. Ruminal fermentation assays showed that 1,3-PDN reduced CH4 production by ~55%, and altered hydrogen (H2) metabolism, leading to 11-fold increase in H2 accumulation. Metatranscriptomic profiling revealed that 1,3-PDN significantly altered the active archaeal community, with a notable reduction in Methanobrevibacter_A and suppression of hydrogenotrophic methanogenesis. Molecular docking suggested that 1,3-PDN may bind to the conserved active site of methyl-coenzyme M reductase (MCR), potentially contributing to MCR-associated inhibition. Proteomic analysis further indicated that 1,3-PDN supplementation downregulated key MCR subunits and simultaneously affected other redox-sensitive methanogenesis-related processes, including tetrahydromethanopterin S-methyltransferase (MTR) subunits and proteins involved in the biosynthesis of cofactors F430 and cobalamin. Nitrogen-equivalent assay suggested partial contribution of nitrite to the methanogenesis inhibition and oxidative effects induced by 1,3-PDN. Together, these findings identify 1,3-PDN as an effective inhibitor of ruminal methanogenesis with enhanced cellular enrichment, providing mechanistic insights for the rational development of new CH4 inhibitors.
|
|
Scooped by
mhryu@live.com
Today, 12:33 AM
|
GapR is a pleiotropic α-proteobacterial nucleoid-associated protein (NAP) reported to either directly regulate transcription of AT-rich DNA or to regulate transcription indirectly through sensing DNA topology and modulating topoisomerase activity. We use single-DNA micromechanics, biolayer interferometry (BLI), and computational analysis to study GapR transcriptional regulation. Micromechanics experiments show that GapR overtwists DNA and shortens its contour length. GapR binding also substantially increases DNA bending persistence length and twist stiffness. For DNA tension ∼0.5 pN, as occurs in supercoiled domains, GapR also promotes DNA strand separation, a transition not observed for lower forces. Nonequilibrium binding experiments show GapR–DNA complexes to be extremely stable, with essentially no dissociation on hour-long time scales. Strikingly, our BLI experiments show that GapR has high affinity for AT-rich DNA, while our micromechanics show that GapR binding is enhanced by pre-twisting of DNA, validating GapR affinity for both forms. By analyzing published GapR binding data, we reveal that overtwisted DNA primarily determines GapR localization. We demonstrate that these two binding modes have opposing impacts on gene expression, with AT-rich binding activating and overtwisted DNA binding repressing transcription. Together, our findings demonstrate how the biophysical activities and topological sensitivity of a single NAP generate context-specific behavior.
|
|
Scooped by
mhryu@live.com
Today, 12:16 AM
|
Three-dimensional structural reconstruction revealed that localized cell wall degradation is essential for the transition from the infection thread (IT) to the infection droplet (ID). Specifically, NPL-mediated local pectin degradation drives this transition and promotes efficient bacterial release.
|
|
Scooped by
mhryu@live.com
July 28, 11:46 PM
|
Recent advances in AI inspire visions of universal models of biology. Yet living systems are evolved, emergent processes whose behaviors cannot be inferred from their parts alone. We propose grounding AI in canonical biological processes, constructing data-driven world models with explicit mechanistic links across molecules, cells, and their dynamics in space and time.
|
|
Scooped by
mhryu@live.com
July 28, 6:31 PM
|
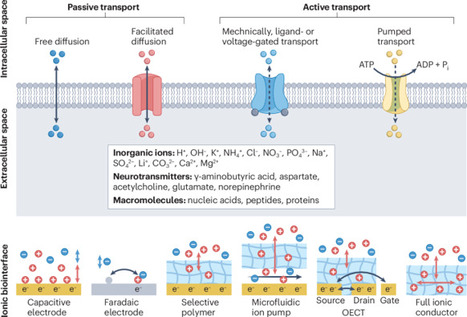
Bioiontronics is an interdisciplinary field emerging from a confluence of breakthroughs in iontronics and bioengineering. Bioiontronics promotes communication with living matter through ions and biological molecules, thereby mediating the detection and modulation of biological activities. This process can function at abiotic–biotic interfaces in an autonomous fashion or in integral, modular components of biomedical devices. The clinical need to use specific biomolecular information, such as ion concentrations and biomarker levels, for personalized diagnostics and treatments has driven the rapid evolution of bioiontronics, through the convergence of fundamental science and application-oriented engineering. However, several challenges remain, including control of ion transport, device miniaturization, biocompatible encapsulation for long-term implantation and tools to decipher and then harness multimodal biological signals. In this Review, we discuss the underlying mechanisms and recent prototypes of bioiontronic devices and describe challenges in the fabrication and application of such systems. Bioiontronics enables communication with living systems through controlled ion and biomolecule transport at abiotic–biotic interfaces. This Review outlines the mechanisms and emerging device prototypes driving the field and discusses key challenges in ion control, miniaturization, biocompatible fabrication, as well as diagnostic and therapeutic applications.
|
|
|
Scooped by
mhryu@live.com
Today, 3:03 PM
|
Global food security is increasingly threatened by climate change, as rising temperatures compromise the yields of major staple crops, including wheat, rice, and maize. Enhancing plant thermotolerance has therefore become a critical priority for sustaining agricultural productivity. However, plant heat-stress responses have often been described as fragmented and pathway-specific, limiting their translation into effective crop improvement strategies. Here, we synthesize current knowledge of plant responses to heat stress and reframe them as an integrated set of design principles centered on preserving photosynthetic carbon gain under high temperature. By doing so, we provide a collection of insights that may help guide future research efforts and the development of strategies for improving plant thermotolerance. We organize major defense strategies into six functional domains: (i) membrane systems and structural integrity, (ii) photosynthetic regulation, (iii) protective metabolites and hormonal signaling, (iv) reactive oxygen species (ROS) scavenging, (v) protein homeostasis, and (vi) transcriptional and post-transcriptional regulation. Rather than treating these responses as independent pathways, we emphasize their temporal hierarchy, energetic costs, and functional interconnections, highlighting their shared objective—maintaining CO2 assimilation, energy balance, and biomass accumulation as thermal damage accelerates. We further discuss how insights from mutagenesis, transgenic approaches, and targeted genetic modification can be translated into crop improvement, clarifying opportunities and trade-offs that emerge when thermotolerance is engineered at distinct physiological nodes. Together, this design-centered framework provides a unifying conceptual and practical roadmap for developing high-yielding, heat-tolerant cultivars, offering actionable guidance for sustaining crop productivity in a warming world.
|
|
Scooped by
mhryu@live.com
Today, 2:59 PM
|
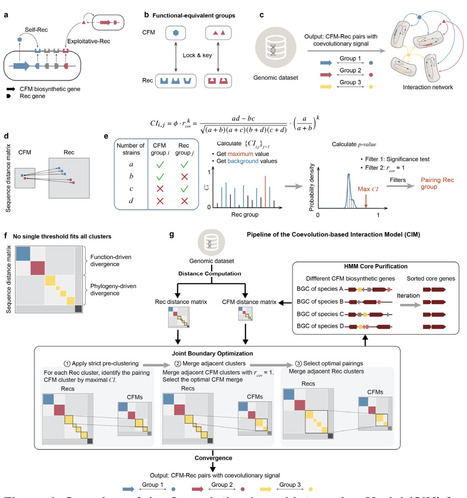
Microbial communities are shaped by secreted metabolites that mediate ecological interactions, yet predicting these interactions from genomic sequences remains difficult, because the specific recognition between co-functional metabolites (CFMs), such as siderophores, and their receptor proteins (Rec) cannot be inferred from gene annotation alone. This difficulty arises from three factors: the prevalence of Rec-mediated exploitation, the lack of high-accuracy functional annotations, and the absence of genomic co-localization between functionally paired CFM-Rec in Gram-positive bacteria. Here we present the Coevolution-based Interaction Model (CIM), an automated framework that maps specific CFM-Rec pairings directly from uncurated genomic datasets. Using a dynamic joint optimization strategy that accounts for exploitation asymmetry and avoids combinatorial explosion, CIM identifies functional pairings solely through evolutionary covariation. We validated this approach by reconstructing macroscale iron scavenging networks across nine bacterial taxa. Experiments confirmed that CIM can bridge genomic distances exceeding 3 Mb in the Gram-positive genus Rhodococcus to identify unlinked cognate receptors, and can accurately predict cross-utilization by exploiter strains despite substantial receptor sequence heterogeneity in Burkholderiaceae and Rhizobiaceae. Finally, topological analysis of the reconstructed networks shows that siderophore exploitation acts as a universal topological glue, fusing fragmented microbial populations into highly connected communities, and that the exploitability of siderophore production reverses depending on network modularity. CIM thus offers a scalable, sequence-to-ecology approach for predicting interactions mediated by secondary metabolites in microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 2:52 PM
|
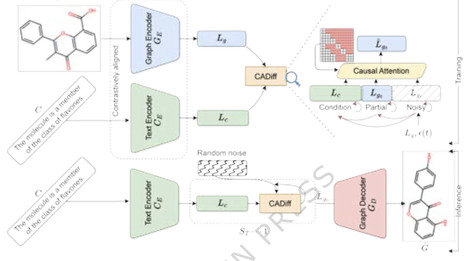
The design of molecules with desired properties is a key challenge in drug discovery and materials science. Traditional methods rely on trial-and-error, while recent deep-learning approaches accelerate molecular generation. However, existing models struggle with generating molecules based on specific textual descriptions. We introduce Mol-CADiff, a diffusion-based framework that uses causal attention mechanisms for text-conditional molecular generation. Our approach explicitly models the causal relationship between textual prompts and molecular structures, overcoming limitations in existing methods. We enhance dependency modeling both within and across modalities, enabling precise control over the generation process. While primarily designed for text-guided tasks, this architecture inherently supports unconditional generation, providing the added capability to autonomously sample the broader chemical space without explicit constraints. Here we show that Mol-CADiff outperforms alternative methods in generating diverse, chemically valid molecules, with better alignment to specified properties, enabling more intuitive language-driven molecular design. By bridging these modalities, our framework provides a versatile method for drug discovery. Computational approaches to molecular design often explore only limited regions of the vast chemical space. This study presents a causality-aware diffusion model that generates valid and diverse molecules with or without text prompts, improving controllability in molecular design.
|
|
Scooped by
mhryu@live.com
Today, 2:46 PM
|
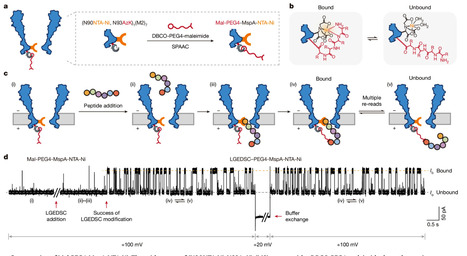
Accurate decoding of peptide sequences is crucial in proteomics. However, achieving this goal is a technical challenge, owing to the compositional and structural complexity of peptides. Studies inspired by the success of nanopore nucleic acid sequencing have shown that nanopore-based techniques can also be applied to peptide sequencing. The key is to generate narrowly distributed, consistent and sequence-dependent events during sequential nanopore readout. Here we introduce a nanopore-based strategy termed transient pore analyte looping (tPAL). We develop an engineered Mycobacterium smegmatis porin A (MspA) nanopore that is dual modified with a nickel-ion-bound nitrilotriacetic acid (NTA-Ni) adapter and the target peptide. This distinctive sensing configuration enables precise recognition of the N terminus of the immobilized peptide by multiple re-readings. With the aid of cholesterolized aminopeptidase, the immobilized peptide can be shortened sequentially in single-amino-acid increments, yielding sequence-dependent, stepwise and narrowly distributed signal alterations that provide clues to allow peptide sequence decoding. Our strategy achieves single-amino-acid resolution and effectively identifies single-amino-acid mutations, post-translational modifications and unnatural-amino-acid insertions—indicative of its versatility in nanopore proteomics and chiral peptide analyses. A nanopore-based ‘chop and measure’ method sequences peptides at single-amino-acid resolution by using enzymatic digestion to progressively shorten the N terminus one residue at a time, together with repetitive N-terminus re-reading.
|
|
Scooped by
mhryu@live.com
Today, 1:23 AM
|
Bacterial–fungal interactions represent fundamental ecological associations that shape microbial community structure across diverse environments. While traditionally framed through the lens of antagonism, bacteria and fungi engage in sophisticated metabolic dialogs extending far beyond simple warfare. Both primary and specialized metabolites function as context-dependent signals, nutrient resources, and modulators of cellular processes that fundamentally influence the physiology, development, and evolutionary trajectory of both fungi and bacteria. Primary metabolites mediate mutualistic relationships through cross-feeding and syntrophy, while specialized metabolites, including volatile organic compounds, lipopeptides, and phenazines, modulate fungal physiology at sub-inhibitory concentrations, reprogramming metabolic networks and triggering adaptive responses without causing cell death. At the molecular level, bacterial metabolites regulate fungal gene expression through transcriptional reprogramming, with consequences that extend to long-term evolutionary adaptation. The complexity of Bacillus–Trichoderma interactions exemplifies how these principles translate into ecological outcomes, exhibiting context-dependent transitions from competition to synergism that enhance biocontrol efficacy, plant growth promotion, and organic matter turnover. Recognizing the multifunctional nature of microbial metabolites beyond direct toxicity opens new avenues for rationally engineering cross-kingdom consortia with targeted agricultural and biotechnological applications.
|
|
Scooped by
mhryu@live.com
Today, 1:19 AM
|
The new laws break away from 20-year-old restrictive GMO directives to give an official nod to plants made with new genomic techniques.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
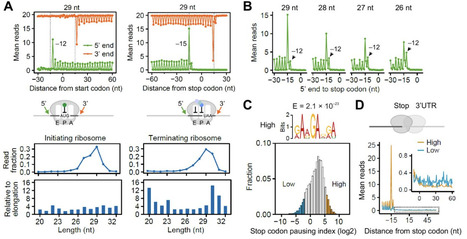
Accurate termination of protein synthesis is paramount for the integrity of the cellular proteome, yet the dynamics and fidelity of ribosome termination remain poorly understood. Here, we establish a profiling strategy to capture terminating ribosomes in mammalian cells and reveal a substantial heterogeneity in ribosome pausing at individual stop codons. We identify a sequence motif upstream of the stop codon that promotes termination pausing, a finding supported by massively parallel reporter assays. Unexpectedly, reduced termination pausing increases the likelihood of stop codon slippage, giving rise to proteins with heterogeneous C-terminal extensions. Mechanistically, we show that sequence-dependent termination pausing is consistent with post-decoding mRNA scanning by the 3′ end of 18 S rRNA. We further uncover tissue-specific patterns of termination pausing that correlate with the stoichiometry of Rps26, which potentially modulates mRNA:rRNA interactions. Together, these results suggest termination pausing as a distinct translational signature shaped by mRNA sequence contexts, ribosome heterogeneity, and cell type-specific translational control.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
Bacterial cellulose (BC) is a biopolymer that comes from natural sources. It has high purity, crystallinity, mechanical strength, and biocompatibility, which makes it suitable for various biomedical and industrial uses. Unlike plant cellulose, BC does not contain lignin or hemicellulose. This results in better material quality and simpler processing. However, producing BC on a large scale is limited by high production costs, expensive culture media, and dependence on a few bacterial strains. It is essential to find cost-effective production methods and alternative microbial sources to expand its commercial use. This study looked at the cellulose-producing ability of Acetobacter diazotrophicus, a safe and relatively unexplored bacterium, under different growth conditions. We compared bacterial growth and cellulose production using Hestrin - Schramm (HS) medium, the standard for BC production, and LB supplemented with glucose (LB+Glucose), which we explored as a more affordable option. We analyzed growth rates, inoculum age, and pH levels to find the best conditions for cellulose production. We observed faster bacterial growth in HS medium, with a doubling time of 3.906 hours, compared to 6.241 hours in LB+Glucose medium. Cellulose production was greatly affected by inoculum age, with successful synthesis from cultures that were agitated for 36 to 40 hours. The highest cellulose yield was at pH 6.0 in HS medium (4.6 mg/mL) and at pH 5.5 in LB+Glucose medium (3.6 mg/mL). FTIR analysis confirmed the presence of characteristic functional groups of bacterial cellulose. These results suggest that Acetobacter diazotrophicus is a promising and cost-effective option for producing bacterial cellulose and highlight the importance of medium composition, inoculum age, and pH for optimizing production.
|
|
Scooped by
mhryu@live.com
Today, 12:46 AM
|
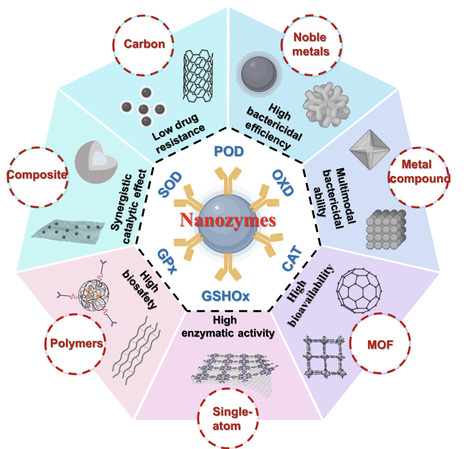
Nanozymes are emerging as versatile antimicrobial agents to tackle drug-resistant infections and biofilm-associated persistence. In this review, we present an integrated framework that links natural enzyme-mediated host defense to the rational development of antimicrobial nanozymes, and summarize recent advances through a mechanism-materials-engineering-application pipeline. We first summarize representative bactericidal principles of natural enzymes to highlight bioinspired catalytic motifs relevant to nanozyme design. We then classify antibacterial nanozymes by catalytic reaction types, including oxidoreductase- and hydrolase-like activities, and by material platforms, highlighting structure–activity relationships that govern catalytic behavior and antimicrobial performance. Building on these mechanistic and structural insights, we summarize engineering-enhanced bactericidal modalities, including photothermal and photodynamic assistance, metal ion release, immune modulation, cascade catalysis, microenvironment-responsive regulation, and targeting, that help overcome constraints imposed by infectious microenvironments, such as hypoxia, limited hydrogen peroxide availability, elevated antioxidant levels, and biofilm barriers. Finally, we translate these principles into application-oriented guidance across interfaces ranging from abiotic surface protection, including antifouling and device coatings, to superficial and deep-seated infections, and we briefly discuss emerging nanozyme strategies for antifungal and antiviral interventions. Throughout, we emphasize translational considerations, such as activity benchmarking, biosafety evaluation, and scalable manufacturing, to support the development of clinically relevant antimicrobial nanozymes.
|
|
Scooped by
mhryu@live.com
Today, 12:17 AM
|
|
|
Scooped by
mhryu@live.com
Today, 12:13 AM
|
Microorganisms dominate life in the hadal zone, yet extreme sampling difficulty and low biomass have precluded characterization of their in situ activities. Here, we analyze microbiome samples collected from hadal seawaters via in situ filtration during 12 human-occupied vehicle dives. DNA-protein co-extraction and metagenome-guided metaproteomic analysis identify 135,073 non-redundant active proteins, with over 95% being hadal-specific. Metaproteomic quantification distinguishes highly active and less active taxa that differ in biogeographic origins and genomic traits. Hadal microorganisms operate a metabolic regime fundamentally distinct from the upper ocean, preferentially utilizing refractory organic matter (aromatics, halogenated compounds, and D-amino acids) and expanded electron acceptors (thiosulfate and heavy metals), collectively shaping hadal element cycling. Active viruses extend beyond “Piggyback-the-Winner” dynamics, enhancing host adaptation through auxiliary metabolic genes. These findings provide proteome-level evidence of hadal microbial activities and reveal biogeochemical cycling distinct from that of the upper ocean, highlighting the underappreciated significance of hadal microbiomes within global ocean ecosystems.
|
|
Scooped by
mhryu@live.com
July 28, 11:45 PM
|
RiPP structural complexity is significantly expanded by multinuclear non-heme iron-dependent oxidative enzymes (MNIOs). Here, we characterize pseudoprobactin 1 and 2, two MNIO-modified proteins from Pseudomonas protegens Pf-5. Using MS, NMR, and X-ray crystallography, we show that the PbnBC converts precursor cysteines into 5-thiooxazoles. While the precursors feature an N-terminal signal peptide and an intramolecular disulfide, both are dispensable for catalysis. Instead, residues downstream of the target cysteines are the primary determinants of substrate recognition. Furthermore, PbnB2C2 modifies multiple sites in a strictly ordered, stepwise manner. Functionally, pseudoprobactins coordinate Cu2+, enhancing bacterial fitness under chlorite-induced oxidative stress. This work establishes 5-thiooxazole as a widespread MNIO-mediated modification, defines its biosynthetic logic, and reveals a role for MNIO-modified proteins in bacterial oxidative stress defense.
|


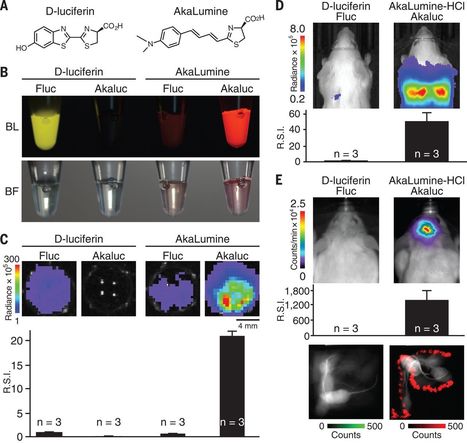



















wonder how this luciferase works in microbes and plants. What is the minimum conc. of AkaLumine (substrate) for luminescence?