Your new post is loading...
Your new post is loading...

|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
March 15, 2012 7:22 AM
|
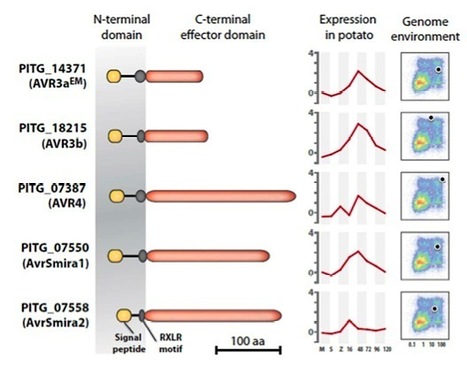
Potato defends against Phytophthora infestans infection by R-gene-based qualitative resistance as well as a quantitative field resistance. R genes are renowned to be rapidly overcome by this oomycete, and potato cultivars with a decent and durable resistance to current P. infestans populations are hardly available. However, potato cultivar Sarpo Mira has retained resistance in the field over several years. We dissected the resistance of cultivar Sarpo Mira in a segregating population by matching the responses to P. infestans RXLR effectors with race-specific resistance to differential strains. The resistance is based on the combination of four pyramided qualitative R genes and a quantitative R gene that was associated with field resistance. The qualitative R genes include R3a, R3b, R4 and the newly identified Rpi-Smira1. The qualitative resistances matched responses to AVR3a, AVR3b, AVR4 and AVRSmira1 RXLR effectors and were overcome by particular P. infestans strains. The quantitative resistance was determined to be conferred by a novel gene Rpi-Smira2. It was only detected under field conditions and was associated with responses to the RXLR effector AvrSmira2. We foresee that effector-based resistance breeding will facilitate selecting and combining qualitative and quantitative resistances that may lead to a more durable resistance to late blight.
|
|
Scooped by
Kamoun Lab @ TSL
January 13, 2012 2:51 AM
|
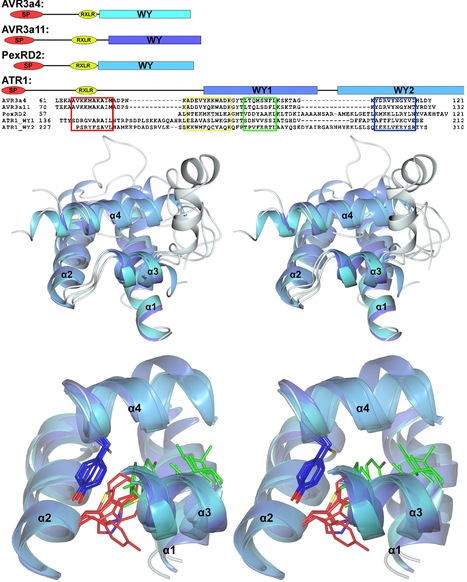
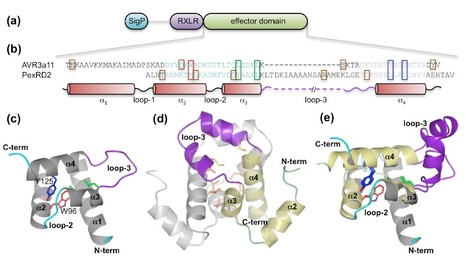
Our laboratories have employed structural biology to investigate the molecular basis of RXLR effector function. A total of four structures have recently been published, those of AVR3a4 and AVR3a11 (paralogues from Phytophthora capsici), PexRD2 (from P. infestans), and ATR1 (from H. arabidopsidis) [21]–[23]. Each publication focused on a different aspect of structure/function analysis including phospholipid binding, protein folding, and effector recognition by the host. The studies of Boutemy et al. and Chou et al. independently described the structural homology of AVR3a11 and a domain of ATR1, respectively, to the cyanobacterial four-helix bundle protein KaiA [24]. This strongly implied they would also be structurally related to each other. This is unexpected, as these Phytophthora and H. arabidopsidis effectors do not share any significant sequence similarity: the conservation was only apparent after the structures were determined and compared.
|
|
Scooped by
Kamoun Lab @ TSL
January 10, 2012 4:32 AM
|
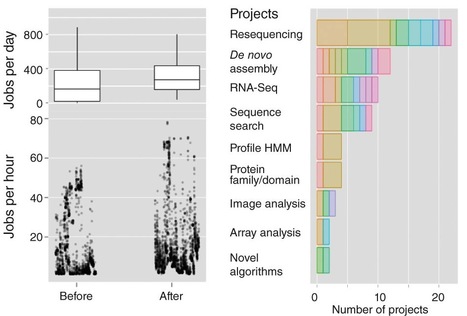
Recently, several articles have focused on the need for flexible, scalable approaches to bioinformatics provision in smaller research institutes and university departments1, 2, 3. As a small institute of around 80 researchers, the Sainsbury Laboratory (Norwich, UK) has been working for the past four years to adapt to the influx of big data sets from high-throughput approaches. In that time, we have successfully transitioned from a 'top-down' model of bioinformatics provision to a 'bottom up' model that incorporates several features discussed in recent articles1, 2, 3. As a result, we have sped up the analysis cycle and can now handle increasing workloads in a timely, productive manner with a modest core support team. Here we provide a description of how we achieved this upgrade in the hope that our experience will prove useful for other small institutions seeking to address the informatics challenges posed by large-scale biological research approaches.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
December 5, 2011 10:00 PM
|
In response to pathogen attack, plant cells secrete antimicrobial molecules at the site of infection. However, how plant pathogens interfere with defense-related focal secretion remains poorly known. Here we show that the host-translocated RXLR-type effector protein AVRblb2 of the Irish potato famine pathogen Phytophthora infestans focally accumulates around haustoria, specialized infection structures that form inside plant cells, and promotes virulence by interfering with the execution of host defenses. AVRblb2 significantly enhances susceptibility of host plants to P. infestans by targeting the host papain-like cysteine protease C14 and specifically preventing its secretion into the apoplast. Plants altered in C14 expression were significantly affected in susceptibility to P. infestans in a manner consistent with a positive role of C14 in plant immunity. Our findings point to a unique counterdefense strategy that plant pathogens use to neutralize secreted host defense proteases. Effectors, such as AVRblb2, can be used as molecular probes to dissect focal immune responses at pathogen penetration sites.
|
|
Scooped by
Kamoun Lab @ TSL
August 3, 2011 10:04 PM
|
Phytopathogens deliver effector proteins inside host plant cells to promote infection. These proteins can also be sensed by the plant immune system, leading to restriction of pathogen growth. Effector genes can display signatures of positive selection and rapid evolution, presumably a consequence of their co-evolutionary arms race with plants. The molecular mechanisms underlying how effectors evolve to gain new virulence functions and/or evade the plant immune system are poorly understood. Here, we report the crystal structures of the effector domains from two oomycete RXLR proteins, Phytophthora capsici AVR3a11 and Phytophthora infestans PexRD2. Despite sharing < 20% sequence identity in their effector domains they display a conserved core α-helical fold. Bioinformatic analyses suggest the core fold occurs in ~44% of annotated Phytophthora RXLR effectors, both as a single domain and in tandem repeats of up to 11 units. Functionally important and polymorphic residues map to the surface of the structures and PexRD2, but not AVR3a11, oligomerizes in planta. We conclude that the core α-helical fold enables functional adaptation of these fast-evolving effectors through (i) insertion/deletions in loop regions between α-helices, (ii) extensions to the N- and C-termini, (iii) amino acid replacements in surface residues, (iv) tandem domain duplications and (v) oligomerization. We hypothesize that the molecular stability provided by this core fold, combined with considerable potential for plasticity, underlies the evolution of effectors that maintain their virulence activities while evading recognition by the plant immune system.
|
|
Scooped by
Kamoun Lab @ TSL
August 1, 2011 9:58 AM
|
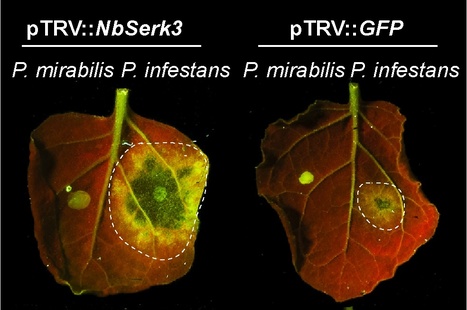
Background - The filamentous oomycete plant pathogen Phytophthora infestans causes late blight, an economically important disease, on members of the nightshade family (Solanaceae), such as the crop plants potato and tomato. The related plant Nicotiana benthamiana is a model system to study plant-pathogen interactions, and the susceptibility of N. benthamiana to Phytophthora species varies from susceptible to resistant. Little is known about the extent to which plant basal immunity, mediated by membrane receptors that recognise conserved pathogen-associated molecular patterns (PAMPs), contributes to P. infestans resistance.
Principal Findings - We found that different species of Phytophthora have varying degrees of virulence on N. benthamiana ranging from avirulence (incompatible interaction) to moderate virulence through to full aggressiveness. The leucine-rich repeat receptor-like kinase (LRR-RLK) BAK1/SERK3 is a major modulator of PAMP-triggered immunity (PTI) in Arabidopsis thaliana and N. benthamiana. We cloned two NbSerk3 homologs, NbSerk3A and NbSerk3B, from N. benthamiana based on sequence similarity to the A. thaliana gene. N. benthamiana plants silenced for NbSerk3 showed markedly enhanced susceptibility to P. infestans infection but were not altered in resistance to Phytophthora mirabilis, a sister species of P. infestans that specializes on a different host plant. Furthermore, silencing of NbSerk3 reduced the cell death response triggered by the INF1, a secreted P. infestans protein with features of PAMPs.
Conclusions/Significance - We demonstrated that N. benthamiana NbSERK3 significantly contributes to resistance to P. infestans and regulates the immune responses triggered by the P. infestans PAMP protein INF1. In the future, the identification of novel surface receptors that associate with NbSERK3A and/or NbSERK3B should lead to the identification of new receptors that mediate recognition of oomycete PAMPs, such as INF1.
|
|
Scooped by
Kamoun Lab @ TSL
August 1, 2011 10:15 AM
|
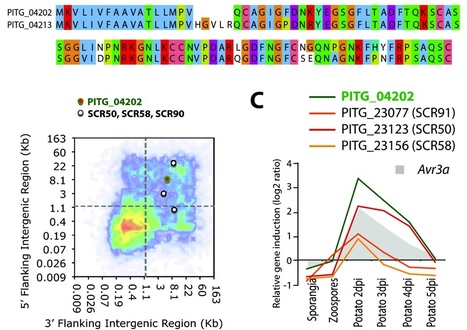
Background - Phytophthora infestans is the most devastating pathogen of potato and a model organism for the oomycetes. It exhibits high evolutionary potential and rapidly adapts to host plants. The P. infestans genome experienced a repeat-driven expansion relative to the genomes of Phytophthora sojae and Phytophthora ramorum and shows a discontinuous distribution of gene density. Effector genes, such as members of the RXLR and Crinkler (CRN) families, localize to expanded, repeat-rich and gene-sparse regions of the genome. This distinct genomic environment is thought to contribute to genome plasticity and host adaptation.
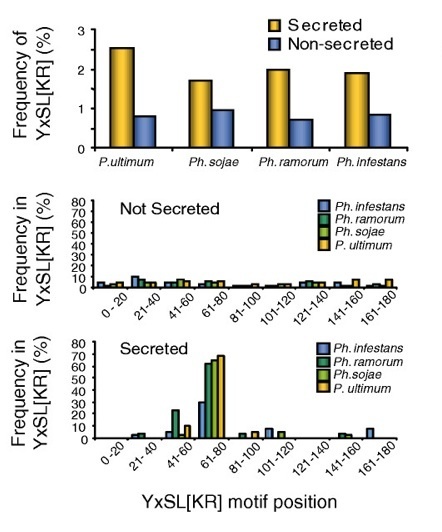
Results - We used in silico approaches to predict and describe the repertoire of P. infestans secreted proteins (the secretome). We defined the "plastic secretome" as a subset of the genome that (i) encodes predicted secreted proteins, (ii) is excluded from genome segments orthologous to the P. sojae and P. ramorum genomes and (iii) is encoded by genes residing in gene sparse regions of P. infestans genome. Although including only ~3% of P. infestans genes, the plastic secretome contains ~62% of known effector genes and shows >2 fold enrichment in genes induced in planta. We highlight 19 plastic secretome genes induced in planta but distinct from previously described effectors. This list includes a trypsin-like serine protease, secreted oxidoreductases, small cysteine-rich proteins and repeat containing proteins that we propose to be novel candidate virulence factors.
Conclusions - This work revealed a remarkably diverse plastic secretome. It illustrates the value of combining genome architecture with comparative genomics to identify novel candidate virulence factors from pathogen genomes.
|
|
Scooped by
Kamoun Lab @ TSL
August 1, 2011 10:33 AM
|
Pathogens use specialized secretion systems and targeting signals to translocate effector proteins inside host cells, a process that is essential for promoting disease and parasitism. However, the amino acid sequences that determine host delivery of eukaryotic pathogen effectors remain mostly unknown. The Crinkler (CRN) proteins of oomycete plant pathogens, such as the Irish potato famine organism Phytophthora infestans, are modular proteins with predicted secretion signals and conserved N-terminal sequence motifs. Here, we provide direct evidence that CRN N termini mediate protein transport into plant cells. CRN host translocation requires a conserved motif that is present in all examined plant pathogenic oomycetes, including the phylogenetically divergent species Aphanomyces euteiches that does not form haustoria, specialized infection structures that have been implicated previously in delivery of effectors. Several distinct CRN C termini localized to plant nuclei and, in the case of CRN8, required nuclear accumulation to induce plant cell death. These results reveal a large family of ubiquitous oomycete effector proteins that target the host nucleus. Oomycetes appear to have acquired the ability to translocate effector proteins inside plant cells relatively early in their evolution and before the emergence of haustoria. Finally, this work further implicates the host nucleus as an important cellular compartment where the fate of plant–microbe interactions is determined.
|
|
|
Scooped by
Kamoun Lab @ TSL
January 26, 2012 3:35 PM
|
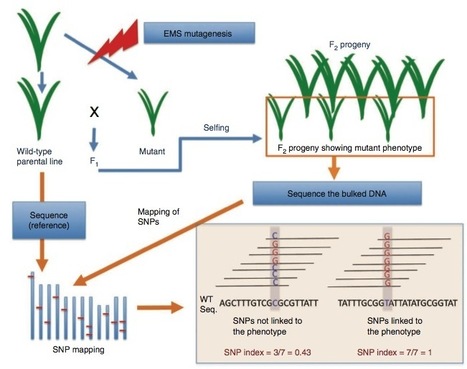
The majority of agronomic traits are controlled by multiple genes that cause minor phenotypic effects, making the identification of these genes difficult. Here we introduce MutMap, a method based on whole-genome resequencing of pooled DNA from a segregating population of plants that show a useful phenotype. In MutMap, a mutant is crossed directly to the original wild-type line and then selfed, allowing unequivocal segregation in second filial generation (F2) progeny of subtle phenotypic differences. This approach is particularly amenable to crop species because it minimizes the number of genetic crosses (n = 1 or 0) and mutant F2 progeny that are required. We applied MutMap to seven mutants of a Japanese elite rice cultivar and identified the unique genomic positions most probable to harbor mutations causing pale green leaves and semidwarfism, an agronomically relevant trait. These results show that MutMap can accelerate the genetic improvement of rice and other crop plants.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
January 10, 2012 10:35 AM
|
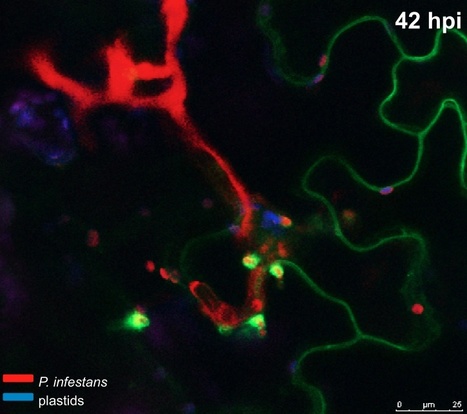
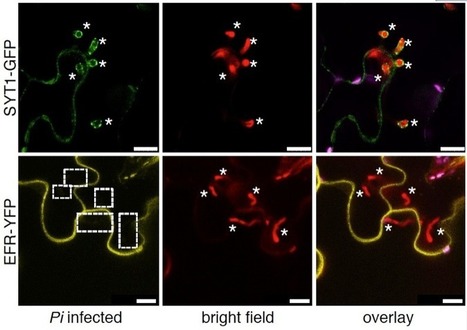
Adapted filamentous pathogens such as the oomycetes Hyaloperonospora arabidopsidis (Hpa) and Phytophthora infestans (Pi) project specialized hyphae, the haustoria, inside living host cells for the suppression of host defense and acquisition of nutrients. Accommodation of haustoria requires reorganization of the host cell and the biogenesis of a novel host cell membrane, the extrahaustorial membrane (EHM), which envelops the haustorium separating the host cell from the pathogen. Here, we applied live-cell imaging of fluorescent-tagged proteins labeling a variety of membrane compartments and investigated the subcellular changes associated with accommodating oomycete haustoria in Arabidopsis and N. benthamiana. Plasma membrane-resident proteins differentially localized to the EHM. Likewise, secretory vesicles and endosomal compartments surrounded Hpa and Pi haustoria revealing differences between these two oomycetes, and suggesting a role for vesicle trafficking pathways for the pathogen-controlled biogenesis of the EHM. The latter is supported by enhanced susceptibility of mutants in endosome-mediated trafficking regulators. These observations point at host subcellular defenses and specialization of the EHM in a pathogen-specific manner. Defense-associated haustorial encasements, a double-layered membrane that grows around mature haustoria, were frequently observed in Hpa interactions. Intriguingly, all tested plant proteins accumulated at Hpa haustorial encasements suggesting the general recruitment of default vesicle trafficking pathways to defend pathogen access. Altogether, our results show common requirements of subcellular changes associated with oomycete biotrophy, and highlight differences between two oomycete pathogens in reprogramming host cell vesicle trafficking for haustoria accommodation. This provides a framework for further dissection of the pathogen-triggered reprogramming of host subcellular changes.
|
|
Scooped by
Kamoun Lab @ TSL
January 7, 2012 10:31 AM
|
Rust fungi are obligate biotrophic pathogens that cause considerable damage on crop plants. Puccinia graminis f. sp. tritici, the causal agent of wheat stem rust, and Melampsora larici-populina, the poplar leaf rust pathogen, have strong deleterious impacts on wheat and poplar wood production, respectively. Filamentous pathogens such as rust fungi secrete molecules called disease effectors that act as modulators of host cell physiology and can suppress or trigger host immunity. Current knowledge on effectors from other filamentous plant pathogens can be exploited for the characterisation of effectors in the genome of recently sequenced rust fungi. We designed a comprehensive in silico analysis pipeline to identify the putative effector repertoire from the genome of two plant pathogenic rust fungi. The pipeline is based on the observation that known effector proteins from filamentous pathogens have at least one of the following properties: (i) contain a secretion signal, (ii) are encoded by in planta induced genes, (iii) have similarity to haustorial proteins, (iv) are small and cysteine rich, (v) contain a known effector motif or a nuclear localization signal, (vi) are encoded by genes with long intergenic regions, (vii) contain internal repeats, and (viii) do not contain PFAM domains, except those associated with pathogenicity. We used Markov clustering and hierarchical clustering to classify protein families of rust pathogens and rank them according to their likelihood of being effectors. Using this approach, we identified eight families of candidate effectors that we consider of high value for functional characterization. This study revealed a diverse set of candidate effectors, including families of haustorial expressed secreted proteins and small cysteine-rich proteins. This comprehensive classification of candidate effectors from these devastating rust pathogens is an initial step towards probing plant germplasm for novel resistance components.
|
|
Scooped by
Kamoun Lab @ TSL
August 5, 2011 6:33 PM
|
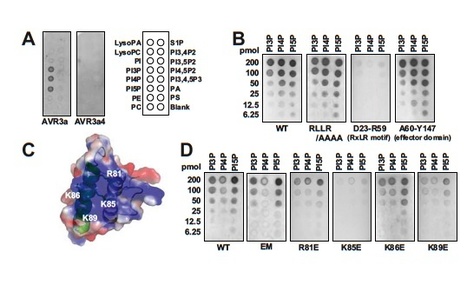
The oomycete pathogen Phytophthora infestans causes potato late blight, one of the most economically damaging plant diseases worldwide. P. infestans produces AVR3a, an essential modular virulence effector with an N-terminal RXLR domain that is required for host-cell entry. In host cells, AVR3a stabilizes and inhibits the function of the E3 ubiquitin ligase CMPG1, a key factor in host immune responses including cell death triggered by the pathogen-derived elicitor protein INF1 elicitin. To elucidate the molecular basis of AVR3a effector function, we determined the structure of Phytophthora capsici AVR3a4, a close homolog of P. infestans AVR3a. Our structural and functional analyses reveal that the effector domain of AVR3a contains a conserved, positively charged patch and that this region, rather than the RXLR domain, is required for binding to phosphatidylinositol monophosphates (PIPs) in vitro. Mutations affecting PIP binding do not abolish AVR3a recognition by the resistance protein R3a but reduce its ability to suppress INF1-triggered cell death in planta. Similarly, stabilization of CMPG1 in planta is diminished by these mutations. The steady-state levels of non–PIP-binding mutant proteins in planta are reduced greatly, although these proteins are stable in vitro. Furthermore, overexpression of a phosphatidylinositol phosphate 5-kinase results in reduction of AVR3a levels in planta. Our results suggest that the PIP-binding ability of the AVR3a effector domain is essential for its accumulation inside host cells to suppress CMPG1-dependent immunity.
|
|
Scooped by
Kamoun Lab @ TSL
August 1, 2011 9:49 AM
|
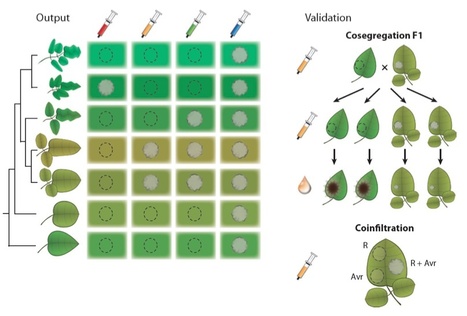
Potato (Solanum tuberosum) is the world’s third-largest food crop. It severely suffers from late blight, a devastating disease caused by Phytophthora infestans. This oomycete pathogen secretes host-translocated RXLR effectors that include avirulence (AVR) proteins, which are targeted by resistance (R) proteins from wild Solanum species. Most Solanum R genes appear to have coevolved with P. infestans at its center of origin in central Mexico. Various R and Avr genes were recently cloned, and here we catalog characterized R-AVR pairs. We describe the mechanisms that P. infestans employs for evading R protein recognition and discuss partial resistance and partial virulence phenotypes in the context of our knowledge of effector diversity and activity. Genome-wide catalogs of P. infestans effectors are available, enabling effectoromics approaches that accelerate R gene cloning and specificity profiling. Engineering R genes with expanded pathogen recognition has also become possible. Importantly, monitoring effector allelic diversity in pathogen populations can assist in R gene deployment in agriculture.
|
|
Scooped by
Kamoun Lab @ TSL
August 1, 2011 10:05 AM
|
Many plant pathogens, including those in the lineage of the Irish potato famine organism Phytophthora infestans, evolve by host jumps followed by specialization. However, how host jumps affect genome evolution remains largely unknown. To determine the patterns of sequence variation in the P. infestans lineage, we resequenced six genomes of four sister species. This revealed uneven evolutionary rates across genomes with genes in repeat-rich regions showing higher rates of structural polymorphisms and positive selection. These loci are enriched in genes induced in planta, implicating host adaptation in genome evolution. Unexpectedly, genes involved in epigenetic processes formed another class of rapidly evolving residents of the gene-sparse regions. These results demonstrate that dynamic repeat-rich genome compartments underpin accelerated gene evolution following host jumps in this pathogen lineage.
|
|
Scooped by
Kamoun Lab @ TSL
August 1, 2011 10:38 AM
|
Background - Pythium ultimum is a ubiquitous oomycete plant pathogen responsible for a variety of diseases on a broad range of crop and ornamental species.
Results - The P. ultimum genome (42.8 Mb) encodes 15,290 genes and has extensive sequence similarity and synteny with related Phytophthora species, including the potato blight pathogen Phytophthora infestans. Whole transcriptome sequencing revealed expression of 86% of genes, with detectable differential expression of suites of genes under abiotic stress and in the presence of a host. The predicted proteome includes a large repertoire of proteins involved in plant pathogen interactions, although, surprisingly, the P. ultimum genome does not encode any classical RXLR effectors and relatively few Crinkler genes in comparison to related phytopathogenic oomycetes. A lower number of enzymes involved in carbohydrate metabolism were present compared to Phytophthora species, with the notable absence of cutinases, suggesting a significant difference in virulence mechanisms between P. ultimum and more host-specific oomycete species. Although we observed a high degree of orthology with Phytophthora genomes, there were novel features of the P. ultimum proteome, including an expansion of genes involved in proteolysis and genes unique to Pythium. We identified a small gene family of cadherins, proteins involved in cell adhesion, the first report of these in a genome outside the metazoans.
Conclusions - Access to the P. ultimum genome has revealed not only core pathogenic mechanisms within the oomycetes but also lineage-specific genes associated with the alternative virulence and lifestyles found within the pythiaceous lineages compared to the Peronosporaceae.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plant Pathogenomics
February 8, 2012 7:51 AM
|
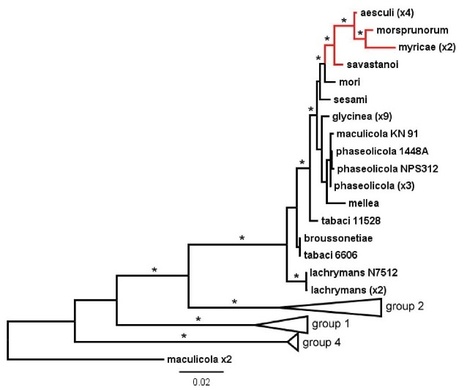
A recently emerging bleeding canker disease, caused by Pseudomonas syringae pathovar aesculi (Pae), is threatening European horse chestnut in northwest Europe. Very little is known about the origin and biology of this new disease. We used the nucleotide sequences of seven commonly used marker genes to investigate the phylogeny of three strains isolated recently from bleeding stem cankers on European horse chestnut in Britain (E-Pae). On the basis of these sequences alone, the E-Pae strains were identical to the Pae type-strain (I-Pae), isolated from leaf spots on Indian horse chestnut in India in 1969. The phylogenetic analyses also showed that Pae belongs to a distinct clade of P. syringae pathovars adapted to woody hosts. We generated genome-wide Illumina sequence data from the three E-Pae strains and one strain of I-Pae. Comparative genomic analyses revealed pathovar-specific genomic regions in Pae potentially implicated in virulence on a tree host, including genes for the catabolism of plant-derived aromatic compounds and enterobactin synthesis. Several gene clusters displayed intra-pathovar variation, including those encoding type IV secretion, a novel fatty acid biosynthesis pathway and a sucrose uptake pathway. Rates of single nucleotide polymorphisms in the four Pae genomes indicate that the three E-Pae strains diverged from each other much more recently than they diverged from I-Pae. The very low genetic diversity among the three geographically distinct E-Pae strains suggests that they originate from a single, recent introduction into Britain, thus highlighting the serious environmental risks posed by the spread of an exotic plant pathogenic bacterium to a new geographic location. The genomic regions in Pae that are absent from other P. syringae pathovars that infect herbaceous hosts may represent candidate genetic adaptations to infection of the woody parts of the tree.
|