 Your new post is loading...
Your new post is loading...

|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
May 29, 2013 8:17 AM
|
|
|
Scooped by
Kamoun Lab @ TSL
April 23, 2013 11:16 AM
|
Background - Wheat yellow (stripe) rust caused by Puccinia striiformis f. sp. tritici (PST) is one of the most devastating diseases of wheat worldwide. To design effective breeding strategies that maximize the potential for durable disease resistance it is important to understand the molecular basis of PST pathogenicity. In particular, the characterisation of the structure, function and evolutionary dynamics of secreted effector proteins that are detected by host immune receptors can help guide and prioritize breeding efforts. However, to date, our knowledge of the effector repertoire of cereal rust pathogens is limited. Results - We re-sequenced genomes of four PST isolates from the US and UK to identify effector candidates and relate them to their distinct virulence profiles. First, we assessed SNP frequencies between all isolates, with heterokaryotic SNPs being over tenfold more frequent (5.29 +/- 2.23 SNPs/kb) than homokaryotic SNPs (0.41 +/- 0.28 SNPs/kb). Next, we implemented a bioinformatics pipeline to integrate genomics, transcriptomics, and effector-focused annotations to identify and classify effector candidates in PST. RNAseq analysis highlighted transcripts encoding secreted proteins that were significantly enriched in haustoria compared to infected tissue. The expression of 22 candidate effector genes was characterised using qRT-PCR, revealing distinct temporal expression patterns during infection in wheat. Lastly, we identified proteins that displayed non-synonymous substitutions specifically between the two UK isolates PST-87/7 and PST-08/21, which differ in virulence to two wheat varieties. By focusing on polymorphic variants enriched in haustoria, we identified five polymorphic effector candidates between PST-87/7 and PST-08/21 among 2,999 secreted proteins. These allelic variants are now a priority for functional validation as virulence/avirulence effectors in the corresponding wheat varieties. Conclusions - Integration of genomics, transcriptomics, and effector-directed annotation of PST isolates has enabled us to move beyond the single isolate-directed catalogues of effector proteins and develop a framework for mining effector proteins in closely related isolates and relate these back to their defined virulence profiles. This should ultimately lead to more comprehensive understanding of the PST pathogenesis system, an important first step towards developing more effective surveillance and management strategies for one of the most devastating pathogens of wheat.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
March 6, 2013 1:05 PM
|
Genome sequences of plant fungal pathogens have enabled the identification of effectors that cooperatively modulate the cellular environment for successful fungal growth and suppress host defense. Identification and characterization of novel effector proteins are crucial to understand pathogen virulence and host plant defense mechanisms. Previous reports indicate that the Pseudomonas syringae pv. tomato DC3000 type III secretion system (T3SS) can be used to study how non-bacterial effectors manipulate dicot plant cell function using the Effector Detector Vector (pEDV) system. Here we report a pEDV-based effector delivery system in which the T3SS of Burkholderia glumae, an emerging rice pathogen, is used to translocate the AVR-Pik and AVR-Pii effectors of fungal pathogen Magnaporthe oryzae to rice cytoplasm. The translocated AVR-Pik and AVR-Pii showed avirulence activity when tested in rice cultivars containing the cognate R genes. AVR-Pik reduced and delayed the hypersensitive response triggered by B. glumae in the non-host plant Nicotiana benthamiana indicative of an immunosuppressive virulence activity. AVR proteins fused with fluorescent protein and nuclear localization signal were delivered by B. glumae T3SS and observed in the nuclei of infected cells in rice, wheat, barley and N. benthamiana. Our bacterial T3SS-enabled eukaryotic effector delivery and subcellular localization assays provide a useful method to identify and study effector functions in monocot plants.
Via Nicolas Denancé, Kamoun Lab @ TSL
|
|
Rescooped by
Kamoun Lab @ TSL
from MutMap
January 12, 2013 3:15 PM
|
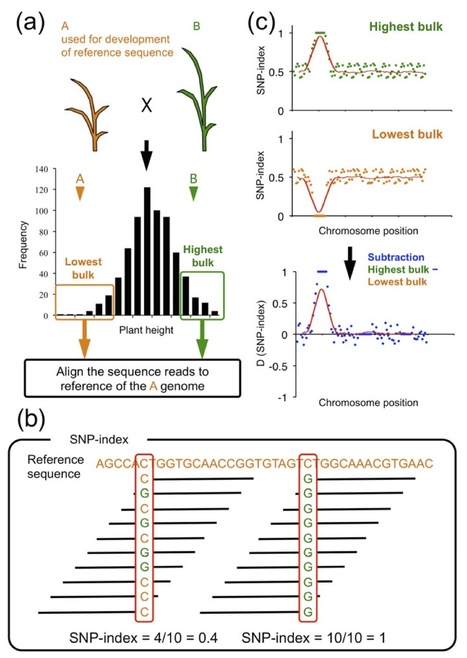
The majority of agronomically important crop traits are quantitative, meaning that they are controlled by multiple genes each with a small effect (quantitative trait loci: QTL). QTL mapping and isolation is important for efficient crop breeding by marker-assisted selection (MAS) and for a better understanding of the molecular mechanisms underlying the traits. Since it requires the development and selection of DNA markers for linkage analysis, QTL analysis has been however time consuming and labor intensive. Here we report a rapid identification of plant QTL by whole genome resequencing of DNAs from two populations each composed of 20-50 individuals showing extreme opposite trait values for a given phenotype in a segregating progeny. We propose to name this approach QTL-seq as applied to plant species. We applied QTL-seq to rice recombinant inbred lines (RILs) and F2 populations and successfully identified QTL for important agronomic traits, such as partial resistance to the fungal rice blast disease and seedling vigor. Simulation study showed that QTL-seq is able to detect QTL over wide ranges of experimental variables, and the method can be generally applied in population genomics studies to rapidly identify genomic regions that underwent artificial or natural selective sweeps.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
December 14, 2012 2:47 AM
|
Welcome to Open Access Data and Crowdsourced analyses!
On this website you'll be able to get data to do your own analyses on ash and ash dieback. You can see the results of other peoples work as soon as it is available and share your own discoveries in the same way.
You will always get full credit for your work and in doing so contribute to a real community effort.
|
|
Scooped by
Kamoun Lab @ TSL
October 25, 2012 9:34 AM
|
Current Biology: A Common Signaling Process that Promotes Mycorrhizal and Oomycete Colonization of Plants (2012)
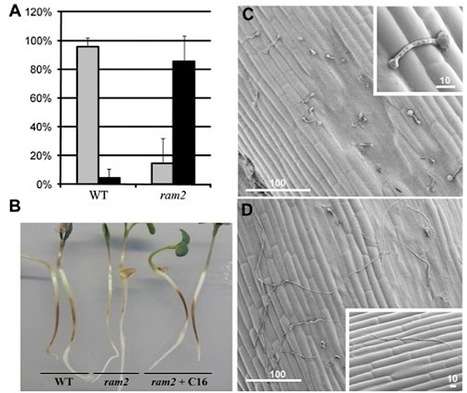
The symbiotic association between plants and arbuscular mycorrhizal fungi is almost ubiquitous within the plant kingdom [1], and the early stages of the association are controlled by plant-derived strigolactone acting as a signal to the fungus in the rhizosphere [2–4] and lipochito-oligosaccharides acting as fungal signals to the plant [5]. Hyphopodia form at the root surface, allowing the initial invasion, and this is analogous to appressoria, infection structures of pathogenic fungi and oomycetes. Here, we characterize RAM2, a gene of Medicago truncatula required for colonization of the root by mycorrhizal fungi, which is necessary for appropriate hyphopodia and arbuscule formation. RAM2 encodes a glycerol-3-phosphate acyl transferase (GPAT) and is involved in the production of cutin monomers. Plants defective in RAM2 are unable to be colonized by arbuscular mycorrhizal fungi but also show defects in colonization by an oomycete pathogen, with the absence of appressoria formation. RAM2 defines a direct signaling function, because exogenous addition of the C16 aliphatic fatty acids associated with cutin are sufficient to promote hyphopodia/ appressoria formation. Thus, cutin monomers act as plant signals that promote colonization by arbuscular mycorrhizal fungi, and this signaling function has been recruited by pathogenic oomycetes to facilitate their own invasion. http://kamounlab.dreamhosters.com/pdfs/CurrBiol_2012.pdf
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
August 23, 2012 5:53 PM
|
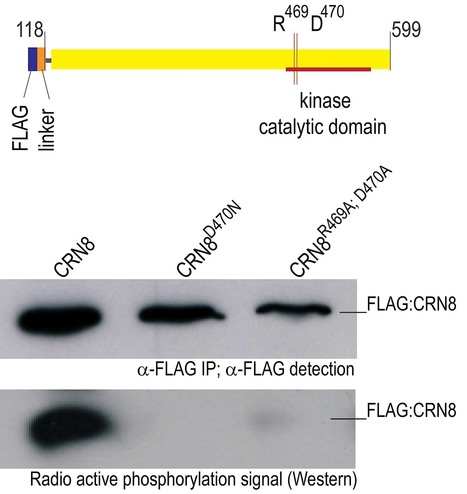
Phytopathogenic oomycetes, such as Phytophthora infestans, secrete an arsenal of effector proteins that modulate plant innate immunity to enable infection. We describe CRN8, a host-translocated effector of P. infestans that has kinase activity in planta. CRN8 is a modular protein of the CRN effector family. The C-terminus of CRN8 localizes to the host nucleus and triggers cell death when the protein is expressed in planta. Cell death induction by CRN8 is dependent on its localization to the plant nucleus, which requires a functional nuclear localization signal (NLS). The C-terminal sequence of CRN8 has similarity to a serine/threonine RD kinase domain. We demonstrated that CRN8 is a functional RD kinase and that its auto-phosphorylation is dependent on an intact catalytic site. Co-immunoprecipitation experiments revealed that CRN8 forms a dimer or multimer. Heterologous expression of CRN8 in planta resulted in enhanced virulence by P. infestans. In contrast, in planta expression of the dominant-negative CRN8R469A;D470A resulted in reduced P. infestans infection, further implicating CRN8 in virulence. Overall, our results indicate that similar to animal parasites, plant pathogens also translocate biochemically active kinase effectors inside host cells.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
July 19, 2012 3:42 AM
|
Every four years, the Olympic Games plays host to competitors who have built on their natural talent by training for many years to become the best in their chosen discipline. Similar spirit and endeavour can be found throughout the microbial world, in which every day is a competition to survive and thrive. Microorganisms are trained through evolution to become the fittest and the best adapted to a particular environmental niche or lifestyle, and to innovate when the 'rules of the game' are changed by alterations to their natural habitats. In this Essay, we honour the best competitors in the microbial world by inviting them to take part in the inaugural Microbial Olympics. Plant pathogens among the Microbial Olympics medalists!
|
|
Scooped by
Kamoun Lab @ TSL
May 17, 2012 7:20 AM
|
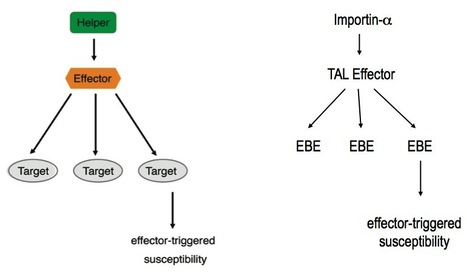
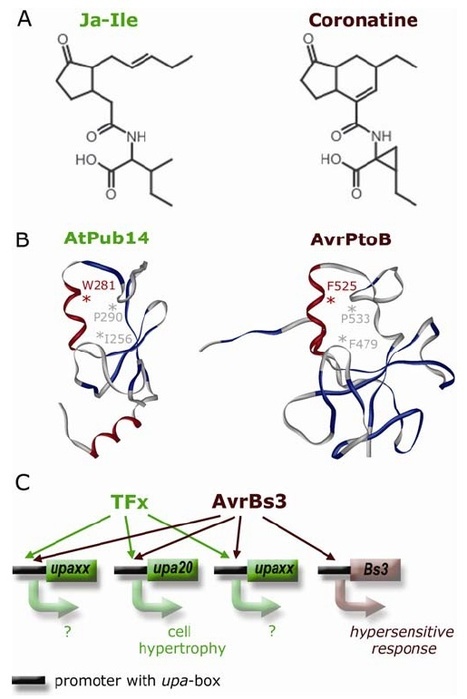
In 2007, Sebastian Schornack, then a freshly minted Ph.D. student from the laboratories of Thomas Lahaye and Ulla Bonas at the Martin-Luther-University Halle-Wittenberg, was fastidiously carrying out follow-up experiments to his thesis work. For the past few years he had been studying how the bacterium Xanthomonas infects its plant hosts. Specifically, he was interested in a class of “effector” proteins, called transcription activator-like (TAL) effectors, that the bacterium delivers to the nuclei of host cells to alter plant gene expression. Ever since their discovery in the late 1980s, the unusual structure of these effector proteins has intrigued plant microbiologists. TAL effectors contain many near-perfect repeats 34 amino acids in length with two hypervariable residues, but the biological meaning of this peculiar modular structure was unknown. At the time Schornack was finishing his thesis, TAL effectors had just been discovered to bind specific DNA sequences in the genomes of their host plants, where they activated expression of host genes thought to favour colonization by the pathogen. While comparing the identity of the hypervariable amino acids in the repeats of particular TAL effectors with the corresponding DNA sequence of their binding sites, Schornack experienced a flash of insight, and noticed a defining pattern. Following discussions with Jens Boch and experimental work with their colleagues at Halle University, it became evident that, indeed, a “code” built into the TAL effector proteins determines their DNA binding specificity. Not long after that, across the Atlantic, another Ph.D. student Matt Moscou, working with Adam Bogdanove at Iowa State University, independently reached a similar conclusion using clever computational analyses of TAL effector-induced expression changes in rice plants. Both teams immediately grasped the impact of their discoveries – synthetic TAL effectors could be custom designed to bind any target DNA sequence. Such a technological breakthrough would have far reaching implications in biotechnology.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
May 10, 2012 6:32 PM
|
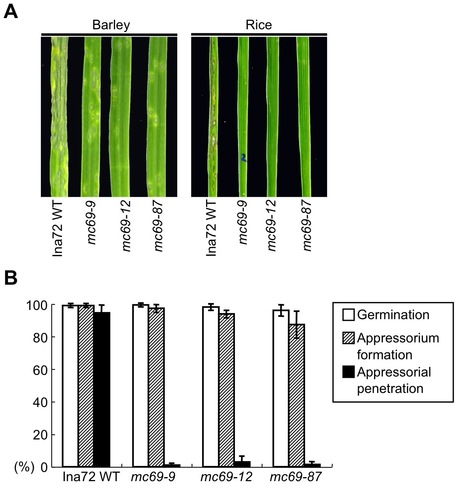
To search for virulence effector genes of the rice blast fungus, Magnaporthe oryzae, we carried out a large-scale targeted disruption of genes for 78 putative secreted proteins that are expressed during the early stages of infection of M. oryzae. Disruption of the majority of genes did not affect growth, conidiation, or pathogenicity of M. oryzae. One exception was the gene MC69. The mc69 mutant showed a severe reduction in blast symptoms on rice and barley, indicating the importance of MC69 for pathogenicity of M. oryzae. The mc69 mutant did not exhibit changes in saprophytic growth and conidiation. Microscopic analysis of infection behavior in the mc69 mutant revealed that MC69 is dispensable for appressorium formation. However, mc69 mutant failed to develop invasive hyphae after appressorium formation in rice leaf sheath, indicating a critical role of MC69 in interaction with host plants. MC69 encodes a hypothetical 54 amino acids protein with a signal peptide. Live-cell imaging suggested that fluorescently labeled MC69 was not translocated into rice cytoplasm. Site-directed mutagenesis of two conserved cysteine residues (Cys36 and Cys46) in the mature MC69 impaired function of MC69 without affecting its secretion, suggesting the importance of the disulfide bond in MC69 pathogenicity function. Furthermore, deletion of the MC69 orthologous gene reduced pathogenicity of the cucumber anthracnose fungus Colletotrichum orbiculare on both cucumber and Nicotiana benthamiana leaves. We conclude that MC69 is a secreted pathogenicity protein commonly required for infection of two different plant pathogenic fungi, M. oryzae and C. orbiculare pathogenic on monocot and dicot plants, respectively.
|
|
Scooped by
Kamoun Lab @ TSL
April 19, 2012 12:30 PM
|
|
|
Scooped by
Kamoun Lab @ TSL
April 8, 2012 10:04 AM
|
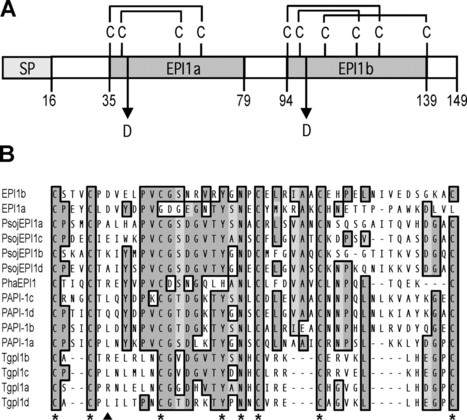
The oomycetes form one of several lineages within the eukaryotes that independently evolved a parasitic lifestyle and consequently are thought to have developed alternative mechanisms of pathogenicity. The oomycete Phytophthora infestans causes late blight, a ravaging disease of potato and tomato. Little is known about processes associated with P. infestans pathogenesis, particularly the suppression of host defense responses. We describe and functionally characterize an extracellular protease inhibitor, EPI1, from P. infestans. EPI1 contains two domains with significant similarity to the Kazal family of serine protease inhibitors. Database searches suggested that Kazal-like proteins are mainly restricted to animals and apicomplexan parasites but appear to be widespread and diverse in the oomycetes. Recombinant EPI1 specifically inhibited subtilisin A among major serine proteases and inhibited and interacted with the pathogenesis-related P69B subtilisin-like serine protease of tomato in intercellular fluids. The epi1 and P69B genes were coordinately expressed and up-regulated during infection of tomato by P. infestans. Inhibition of tomato proteases by EPI1 could form a novel type of defense-counterdefense mechanism between plants and microbial pathogens. In addition, this study points to a common virulence strategy between the oomycete plant pathogen P. infestans and several mammalian parasites, such as the apicomplexan Toxoplasma gondii.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
January 3, 2013 6:59 PM
|
Post-conference thoughts on #OMGN12 Oomycete Molecular Genetics Network Annual Meeting, Nanjing, China
The conference was very much a success. There was notable progress relative to the 2011 Asilomar meeting. Newcomers to the field gave their first OMGN talks and the presentations by local scientists illustrated the wide-range of research taking place in China. I was sorry to miss the closing dinner but I suppose I saved myself the embarrassment of participating in the Karaoke. Below is an attempt to summarize and organize the somewhat random thoughts running through my head right now as I am sitting tight on the Shanghai to New York flight. Thanks to China Eastern Airlines for providing electrical outlets to all economy class passengers. The hot topic: epigenetics - The talks by Wenbo Ma and Mark Gijzen were the most thought provoking for me. Many questions are racing through my head as I am revisiting Wenbo’s talk. To what extent do oomycete effectors suppress RNA silencing? Did they evolve to directly target RNA silencing machinery? What about the nuclear-localized CRNs? Dinah Qutob and Mark Gijzen’s findings also raise many questions. What proportion of Phytophthora genes are targeted by silencing? How frequently do epialleles emerge? How frequently do they revert? What I found particularly exciting about both presentations are the questions they provoke. On the upswing: cell biology - Several presentations included a significant cell biology component but there is room for more. I expect cell biology to become increasingly integrated into oomycete research as we start considering the spatiotemporal aspects of the processes we study. As this happens we need to ensure that we use consistent standards and nomenclature. Not much new: genomics - I did not pick up any obvious new trends in genomics. The loss of heterozygosity (LOH) phenomenon described in a poster by Kurt Lamour et al. is probably the main novelty. There were some new genomes, e.g. Saprolegnia parasitica, Pythium insidiosum, and Pseudoperonospora cubensis. But some of these were described to some extent at previous OMGN conferences. I felt the topic was less dominant than in the past. The talk by the BGI representative was disappointing. What about apoplastic effectors? - How come so few are working on apoplastic effectors? None of the talks addressed these important players in oomycete-plant interactions. Mechanisms of RXLR effector translocation: the plot thickens - There is definitely more clarity than one year ago but many questions remain. The C-terminal binding of AVR3a/AVR1b to PI3Ps described by Yaeno et al. (2011) has been confirmed by Tyler at al. At least this one aspect does not seem to be controversial. But even if AVR3a/AVR1b C-terminal binding to PIPs mediates host cell translocation, it cannot be a general mechanism given that the C-termini of other RXLR effectors do not bind PIPs (I think but I am not sure that this point is accepted by several groups). Kasturi Haldar had some useful advice by reminding us that there are multiple routes to host cell entry in plasmodia. Is the RXLR leader generally involved in PIP binding? - A hotly debated issue remains the lack of reproducibility of the RXLR motif/domain binding to PIPs (Kale et al. 2010 vs. Yaeno et al. 2011 and unpublished reports). The first case of independent confirmation of this central finding of the Kale et al. paper is by Kasturi Haldar’s lab, which showed that the RXLR domain of P. infestans Nuk10 binds PI3Ps. However, the Haldar lab did not test the RXLR leader of AVR3a/AVR1b (pers. comm.), which are the leaders that could not be confirmed by Yaeno et al. and the van West lab (presented at OMGN11). Note also that the Nuk10 leader was not tested by Kale et al. Thus, in my view, the key finding of the Kale et al. paper - that the RXLR leader generally functions in PIP binding – still requires independent confirmation and would be nullified if indeed the AVR3a/AVR1b leader turns out to lack binding activity. What about Kale et al. experiments with full-length effectors? - The validated finding that the C-termini of AVR1b and related effectors bind PIPs compromises a number of experiments described in the Kale et al. paper. Assays in which RXLR mutations in full-length effectors abolish PI3P binding, as well as translocation into cells and roots, are inconsistent with the strong PIP binding of the C-termini. This casts an additional shadow on the quality of the Kale et al. experiments, which have already been plagued by the duplicated figures issue and lack of reproducibility of key assays with the fungal “RXLR-like” sequences. Other host-translocation leaders to the rescue? - Perhaps the breakthrough in understanding how oomycete effectors translocate inside host cells will come from other leaders than RXLR. Lobach, Wawra, van West et al. proposed that binding to tyrosine-O-sulphate on the host cell surface mediates entry of some Saprolegnia parasitica effectors. As far as I can tell this is not controversial although the finding has not been independently confirmed. Still, there is no work reported on other non-RXLR leaders, such as the Crinkler LxLFLAK and Albugo’s CHXC. Lets hope we’ll hear more about these in the future. Pre-secretion sorting? - I was impressed by the talk of Zhijian Zhao, which I thought was the most interesting of the several talks by members of the Nanjing Agricultural University oomycete group. Zhijian is investigating how oomycete effectors are secreted, focusing on the pathogen side. We’ll surely hear more on this topic in the future.
|
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
May 21, 2013 8:16 AM
|
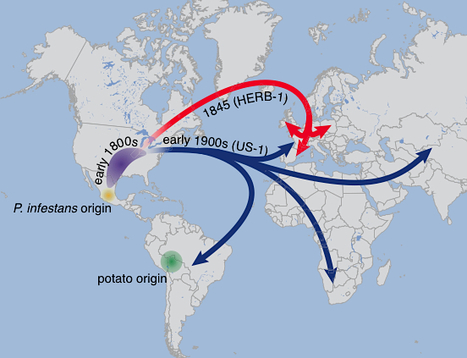
eLife: The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine (2013)
Phytophthora infestans, the cause of potato late blight, is infamous for having triggered the Irish Great Famine in the 1840s. Until the late 1970s, P. infestans diversity outside of its Mexican center of origin was low, and one scenario held that a single strain, US-1, had dominated the global population for 150 years; this was later challenged based on DNA analysis of historical herbarium specimens. We have compared the genomes of 11 herbarium and 15 modern strains. We conclude that the nineteenth century epidemic was caused by a unique genotype, HERB-1, that persisted for over 50 years. HERB-1 is distinct from all examined modern strains, but it is a close relative of US-1, which replaced it outside of Mexico in the twentieth century. We propose that HERB-1 and US-1 emerged from a metapopulation that was established in the early 1800s outside of the species' center of diversity. eLife http://elife.elifesciences.org/content/2/e00731 arXiv preprint http://arxiv.org/abs/1305.4206
|
|
Scooped by
Kamoun Lab @ TSL
March 8, 2013 4:51 AM
|
Listen to Diane Saunders speak about ash fungus genomics and crowdsourcing on BBC Radio 4. See also BBC News - Ash fungus genetic code unravelled http://bbc.in/ZkqY1N
|
|
Rescooped by
Kamoun Lab @ TSL
from Plant Pathogenomics
February 13, 2013 4:41 AM
|
Ash dieback is a devastating fungal disease of ash trees that has swept across Europe and recently reached the UK. This emergent pathogen has received little study in the past and its effect threatens to overwhelm the ash populations. In response to this we have produced some initial genomics datasets and taken the unusual step of releasing them to the scientific for analysis without first performing our own. In this manner we hope to 'crowdsource' analyses and bring the expertise of the community to bear on this problem as quickly as possible. Our data has been released through our website at oadb.tsl.ac.uk and a public GitHub repository.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
December 16, 2012 12:42 PM
|
Chalara ash dieback was first confirmed in the natural environment in the UK in late autumn based on samples from Ashwellthorpe Wood near Norwich. We decided early on in this project that speed would be a critical driver given the emergency nature of the problem. We decided that we should generate genetic sequences as rapidly as possible, release them to the community, and prompt the crowdsourcing exercise we have been publicizing since Friday.
The normal procedure would be to culture the pathogen and sequence the genome and transcriptome from cultured material and controlled laboratory infections. Here, we decided to take the unusual step of directly sequencing the “interaction transcriptome” of a lesion dissected from an infected ash twig. This was the most rapid way to proceed to generate useful information without proceeding through standard laboratory culturing. This is the shortest route from the wood to the sequencer to the computer. The question that many of you must be asking is how useful is this data? This post addresses this question and summarizes the preliminary analyses that the TSL team has produced.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
November 29, 2012 8:54 AM
|
Cold Spring Harbor Symposium on Quantitative Biology: Effector Biology of Plant-Associated Organisms: Concepts and Perspectives (2012)
Every plant is closely associated with a variety of living organisms. Therefore, deciphering howplants interact with mutualistic and parasitic organisms is essential for a comprehensive understanding of the biology of plants. The field of plant–biotic interactions has recently coalesced around an integrated model. Major classes of molecular players both from plants and their associated organisms have been revealed. These include cell surface and intracellular immune receptors of plants, as well as apoplastic and host-cell translocated (cytoplasmic) effectors of the invading organism. This article focuses on effectors, molecules secreted by plant-associated organisms that alter plant processes. Effectors have emerged as a central class of molecules in our integrated view of plant–microbe interactions. Their study has significantly contributed to advancing our knowledge of plant hormones, plant development, plant receptors, and epigenetics. Many pathogen effectors are extraordinary examples of biological innovation; they include some of themost remarkable proteins knownto function inside plant cells. Here, we review some of the key concepts that have emerged from the study of the effectors of plant-associated organisms. In particular, we focus on how effectors function in plant tissues and discuss future perspectives in the field of effector biology. http://kamounlab.dreamhosters.com/pdfs/CSHSQB_2012.pdf
Pest and pathogen losses jeopardise global food security and ever since the 19th century Irish famine, potato late blight has exemplified this threat. The causal oomycete pathogen, Phytophthora infestans, undergoes major population shifts in agricultural systems via the successive emergence and migration of asexual lineages. The phenotypic and genotypic bases of these selective sweeps are largely unknown but management strategies need to adapt to reflect the changing pathogen population. Here, we used molecular markers to document the emergence of a lineage, termed 13_A2, in the European P. infestans population, and its rapid displacement of other lineages to exceed 75% of the pathogen population across Great Britain in less than three years. We show that isolates of the 13_A2 lineage are among the most aggressive on cultivated potatoes, outcompete other aggressive lineages in the field, and overcome previously effective forms of plant host resistance. Genome analyses of a 13_A2 isolate revealed extensive genetic and expression polymorphisms particularly in effector genes. Copy number variations, gene gains and losses, amino-acid replacements and changes in expression patterns of disease effector genes within the 13_A2 isolate likely contribute to enhanced virulence and aggressiveness to drive this population displacement. Importantly, 13_A2 isolates carry intact and in planta induced Avrblb1, Avrblb2 and Avrvnt1 effector genes that trigger resistance in potato lines carrying the corresponding R immune receptor genes Rpi-blb1, Rpi-blb2, and Rpi-vnt1.1. These findings point towards a strategy for deploying genetic resistance to mitigate the impact of the 13_A2 lineage and illustrate how pathogen population monitoring, combined with genome analysis, informs the management of devastating disease epidemics.
Via Nicolas Denancé
|
|
Scooped by
Kamoun Lab @ TSL
August 10, 2012 7:19 PM
|
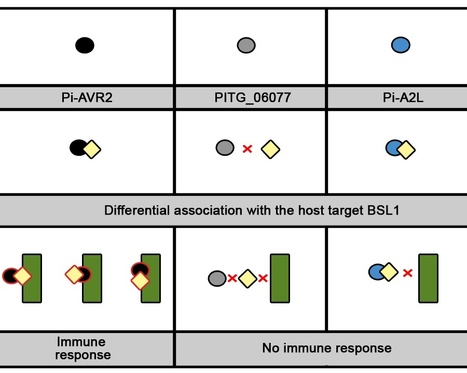
Plant pathogens secrete effector proteins to modulate plant immunity and promote host colonization. Plant nucleotide binding leucine-rich repeat (NB-LRR) immunoreceptors recognize specific pathogen effectors directly or indirectly. Little is known about how NB-LRR proteins recognize effectors of filamentous plant pathogens, such as Phytophthora infestans. AVR2 belongs to a family of 13 sequence-divergent P. infestans RXLR effectors that are differentially recognized by members of the R2 NB-LRR family in Solanum demissum. We report that the putative plant phosphatase BSU-LIKE PROTEIN1 (BSL1) is required for R2-mediated perception of AVR2 and resistance to P. infestans. AVR2 associates with BSL1 and mediates the interaction of BSL1 with R2 in planta, possibly through the formation of a ternary complex. Strains of P. infestans that are virulent on R2 potatoes express an unrecognized form, Avr2-like (referred to as A2l). A2L can still interact with BSL1 but does not promote the association of BSL1 with R2. Our findings show that recognition of the P. infestans AVR2 effector by the NB-LRR protein R2 requires the putative phosphatase BSL1. This reveals that, similar to effectors of phytopathogenic bacteria, recognition of filamentous pathogen effectors can be mediated via a host protein that interacts with both the effector and the NB-LRR immunoreceptor.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plant Pathogenomics
June 21, 2012 12:51 PM
|
The oomycete vegetable pathogen Phytophthora capsici has shown remarkable adaptation to fungicides and new hosts. Like other members of this destructive genus, P. capsici has an explosive epidemiology, rapidly producing massive numbers of asexual spores on infected hosts. In addition, P. capsici can remain dormant for years as sexually-recombined oospores, making it difficult to produce crops at infested sites, and allowing outcrossing populations to maintain significant genetic variation. Genome sequencing, development of a high-density genetic map, and integrative genomic/genetic characterization of P. capsici field isolates and intercross progeny revealed significant mitotic loss of heterozygosity (LOH) and higher levels of single nucleotide variants (SNVs) than those reported for humans, plants, and P. infestans. LOH was detected in clonally propagated field isolates and sexual progeny, cumulatively affecting >30% of the genome. LOH altered genotypes for more than 11,000 SNV sites and showed a strong association with changes in mating type and pathogenicity. Overall, it appears that LOH may provide a rapid mechanism for fixing alleles and may be an important component of adaptability for P. capsici.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plant Pathogenomics
May 17, 2012 7:03 AM
|
Microbiology Today: Genomics of emerging plant pathogens: too little, too late (2012)
Full article at http://www.sgm.ac.uk/pubs/micro_today/pdf/051214.pdf Last year during a visit to Colombia’s Zona Cafetera, my host singled out one coffee farm amid the enchanting rolling hills. That farm’s owner may have looked like the iconic Juan Valdez, but he is far from being cherished by his ‘cafeteros’ colleagues. He is infamous for having brought into Colombia a few coffee plants from Brazil. Unbeknown to him, a few leaves bore small orange spots, the telltale sign of the terrible coffee rust fungus, Hemileia vastatrix. Ever since that fateful introduction in 1983, Colombian cafeteros have struggled with managing this formidable foe. In recent years, after a brief lull, coffee rust came back with a vengeance casting a shadow on a critical Colombian agroindustry just as the country was emerging from years of social instability.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plant Pathogenomics
May 8, 2012 6:05 AM
|
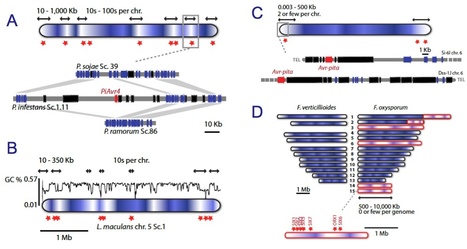
Many species of fungi and oomycetes are plant pathogens of great economic importance. Over the past 7 years, the genomes of more than 30 of these filamentous plant pathogens have been sequenced, revealing remarkable diversity in genome size and architecture. Whereas the genomes of many parasites and bacterial symbionts have been reduced over time, the genomes of several lineages of filamentous plant pathogens have been shaped by repeat-driven expansions. In these lineages, the genes encoding proteins involved in host interactions are frequently polymorphic and reside within repeat-rich regions of the genome. Here, we review the properties of these adaptable genome regions and the mechanisms underlying their plasticity, and we illustrate cases in which genome plasticity has contributed to the emergence of new virulence traits. We also discuss how genome expansions may have had an impact on the co-evolutionary conflict between these filamentous plant pathogens and their hosts.
|
|
Scooped by
Kamoun Lab @ TSL
April 19, 2012 12:50 PM
|
Plant-associated organisms secrete proteins and other molecules to modulate plant defense circuitry and enable colonization of plant tissue. Understanding the molecular function of these secreted molecules, collectively known as effectors, became widely accepted as essential for a mechanistic understanding of the processes underlying plant colonization. This review summarizes recent findings in the field of effector biology and highlights the common concepts that have emerged from the study of cellular plant pathogen effectors.
|
|
Scooped by
Kamoun Lab @ TSL
April 5, 2012 4:02 AM
|
Plant pathogenic oomycetes secrete a diverse repertoire of effector proteins that modulate host innate immunity and enable parasitic infection. Understanding how effectors evolve, translocate and traffic inside host cells, and perturb host processes are major themes in the study of oomycete–plant interactions. The last year has seen important progress in the study of oomycete effectors with, notably, the elucidation of the 3D structures of five RXLR effectors, and novel insights into how cytoplasmic effectors subvert host cells. In this review, we discuss these and other recent advances and highlight the most important open questions in oomycete effector biology.
|




























npr
Scientists can even help history out.