Your new post is loading...
Your new post is loading...

|
Scooped by
Kamoun Lab @ TSL
December 8, 2012 6:58 AM
|
The class Dothideomycetes is one of the largest groups of fungi with a high level of ecological diversity including many plant pathogens infecting a broad range of hosts. Here, we compare genome features of 18 members of this class, including 6 necrotrophs, 9 (hemi)biotrophs and 3 saprotrophs, to analyze genome structure, evolution, and the diverse strategies of pathogenesis. The Dothideomycetes most likely evolved from a common ancestor more than 280 million years ago. The 18 genome sequences differ dramatically in size due to variation in repetitive content, but show much less variation in number of (core) genes. Gene order appears to have been rearranged mostly within chromosomal boundaries by multiple inversions, in extant genomes frequently demarcated by adjacent simple repeats. Several Dothideomycetes contain one or more gene-poor, transposable element (TE)-rich putatively dispensable chromosomes of unknown function. The 18 Dothideomycetes offer an extensive catalogue of genes involved in cellulose degradation, proteolysis, secondary metabolism, and cysteine-rich small secreted proteins. Ancestors of the two major orders of plant pathogens in the Dothideomycetes, the Capnodiales and Pleosporales, may have had different modes of pathogenesis, with the former having fewer of these genes than the latter. Many of these genes are enriched in proximity to transposable elements, suggesting faster evolution because of the effects of repeat induced point (RIP) mutations. A syntenic block of genes, including oxidoreductases, is conserved in most Dothideomycetes and upregulated during infection in L. maculans, suggesting a possible function in response to oxidative stress.
|
|
Scooped by
Kamoun Lab @ TSL
December 2, 2012 4:36 AM
|
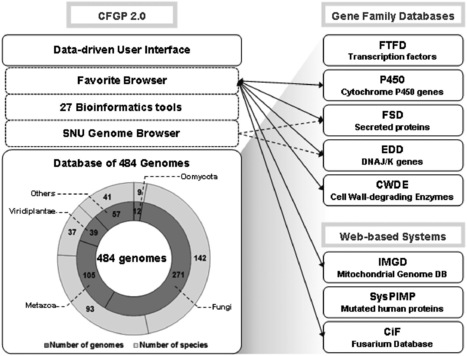
In 2007, Comparative Fungal Genomics Platform (CFGP; http://cfgp.snu.ac.kr/) was publicly open with 65 genomes corresponding to 58 fungal and Oomycete species. The CFGP provided six bioinformatics tools, including a novel tool entitled BLASTMatrix that enables search homologous genes to queries in multiple species simultaneously. CFGP also introduced Favorite, a personalized virtual space for data storage and analysis with these six tools. Since 2007, CFGP has grown to archive 283 genomes corresponding to 152 fungal and Oomycete species as well as 201 genomes that correspond to seven bacteria, 39 plants and 105 animals. In addition, the number of tools in Favorite increased to 27. The Taxonomy Browser of CFGP 2.0 allows users to interactively navigate through a large number of genomes according to their taxonomic positions. The user interface of BLASTMatrix was also improved to facilitate subsequent analyses of retrieved data. A newly developed genome browser, Seoul National University Genome Browser (SNUGB), was integrated into CFGP 2.0 to support graphical presentation of diverse genomic contexts. Based on the standardized genome warehouse of CFGP 2.0, several systematic platforms designed to support studies on selected gene families have been developed. Most of them are connected through Favorite to allow of sharing data across the platforms.
|
|
Scooped by
Kamoun Lab @ TSL
November 9, 2012 10:00 AM
|
Modern agriculture favours the selection and spread of novel plant diseases. Furthermore, crop genetic resistance against pathogens is often rendered ineffective within a few years of its commercial deployment. Leptosphaeria maculans, the cause of phoma stem canker of oilseed rape, develops gene-for-gene interactions with its host plant, and has a high evolutionary potential to render ineffective novel sources of resistance in crops. Here, we established a four-year field experiment to monitor the evolution of populations confronted with the newly released Rlm7 resistance and to investigate the nature of the mutations responsible for virulence against Rlm7. A total of 2551 fungal isolates were collected from experimental crops of a Rlm7 cultivar or a cultivar without Rlm7. All isolates were phenotyped for virulence and a subset was genotyped with neutral genetic markers. Virulent isolates were investigated for molecular events at the AvrLm4-7 locus. Whilst virulent isolates were not found in neighbouring crops, their frequency had reached 36% in the experimental field after four years. An extreme diversity of independent molecular events leading to virulence was identified in populations, with large-scale Repeat Induced Point mutations or complete deletion of AvrLm4-7 being the most frequent. Our data suggest that increased mutability of fungal genes involved in the interactions with plants is directly related to their genomic environment and reproductive system. Thus, rapid allelic diversification of avirulence genes can be generated in L. maculans populations in a single field provided that large population sizes and sexual reproduction are favoured by agricultural practices.
|
|
Scooped by
Kamoun Lab @ TSL
October 7, 2012 1:20 PM
|
Genome sequencing has been carried out on a small selection of major fungal ascomycete pathogens. These studies show that simple models whereby pathogens evolved from phylogenetically related saprobes by the acquisition or modification of a small number of key genes cannot be sustained.The genomes show that pathogens cannot be divided into three clearly delineated classes (biotrophs, hemibiotrophs and necrotrophs) but rather into a complex matrix of categories each with subtly different properties. It is clear that the evolution of pathogenicity is ancient, rapid and ongoing. Fungal pathogens have undergone substantial genomic rearrangements that can be appropriately described as ‘genomic tillage’. Genomic tillage underpins the evolution and expression of large families of genes – known as effectors – that manipulate and exploit metabolic and defence processes of plants so as to allow the proliferation of pathogens.
|
|
Rescooped by
Kamoun Lab @ TSL
from Publications
October 5, 2012 7:53 AM
|
Pest and pathogen losses jeopardise global food security and ever since the 19th century Irish famine, potato late blight has exemplified this threat. The causal oomycete pathogen, Phytophthora infestans, undergoes major population shifts in agricultural systems via the successive emergence and migration of asexual lineages. The phenotypic and genotypic bases of these selective sweeps are largely unknown but management strategies need to adapt to reflect the changing pathogen population. Here, we used molecular markers to document the emergence of a lineage, termed 13_A2, in the European P. infestans population, and its rapid displacement of other lineages to exceed 75% of the pathogen population across Great Britain in less than three years. We show that isolates of the 13_A2 lineage are among the most aggressive on cultivated potatoes, outcompete other aggressive lineages in the field, and overcome previously effective forms of plant host resistance. Genome analyses of a 13_A2 isolate revealed extensive genetic and expression polymorphisms particularly in effector genes. Copy number variations, gene gains and losses, amino-acid replacements and changes in expression patterns of disease effector genes within the 13_A2 isolate likely contribute to enhanced virulence and aggressiveness to drive this population displacement. Importantly, 13_A2 isolates carry intact and in planta induced Avrblb1, Avrblb2 and Avrvnt1 effector genes that trigger resistance in potato lines carrying the corresponding R immune receptor genes Rpi-blb1, Rpi-blb2, and Rpi-vnt1.1. These findings point towards a strategy for deploying genetic resistance to mitigate the impact of the 13_A2 lineage and illustrate how pathogen population monitoring, combined with genome analysis, informs the management of devastating disease epidemics.
Via Nicolas Denancé, Kamoun Lab @ TSL
|
|
Scooped by
Kamoun Lab @ TSL
October 1, 2012 2:27 AM
|
Comparative analyses of pathogen genomes provide new insights into how pathogens have evolved common and divergent virulence strategies to invade related plant species. Fusarium crown and root rots are important diseases of wheat and barley world-wide. In Australia, these diseases are primarily caused by the fungal pathogen Fusarium pseudograminearum. Comparative genomic analyses showed that the F. pseudograminearum genome encodes proteins that are present in other fungal pathogens of cereals but absent in non-cereal pathogens. In some cases, these cereal pathogen specific genes were also found in bacteria associated with plants. Phylogenetic analysis of selected F. pseudograminearum genes supported the hypothesis of horizontal gene transfer into diverse cereal pathogens. Two horizontally acquired genes with no previously known role in fungal pathogenesis were studied functionally via gene knockout methods and shown to significantly affect virulence of F. pseudograminearum on the cereal hosts wheat and barley. Our results indicate using comparative genomics to identify genes specific to pathogens of related hosts reveals novel virulence genes and illustrates the importance of horizontal gene transfer in the evolution of plant infecting fungal pathogens.
|
|
Scooped by
Kamoun Lab @ TSL
September 16, 2012 8:55 AM
|
|
|
Scooped by
Kamoun Lab @ TSL
August 29, 2012 5:03 AM
|
The eukaryotic microbes known as oomycetes are common inhabitants of terrestrial and aquatic environments, and include saprophytes and pathogens. Lifestyles of the pathogens extend from biotrophy to necrotrophy, obligate to facultative pathogenesis, and narrow to broad host ranges on plants or animals. Sequencing of several pathogens has revealed striking variation in genome size and content, a plastic set of genes related to pathogenesis, and adaptations associated with obligate biotrophy. Features of genome evolution include repeat-driven expansions, deletions, gene fusions, and horizontal gene transfer, in a landscape organized into gene-dense and gene-sparse sectors and influenced by transposable elements. Gene expression profiles are also highly dynamic throughout oomycete life cycles, with transcriptional polymorphisms as well as differences in protein sequence contributing to variation. The genome projects have set the foundation for functional studies and should spur the sequencing of additional species, including more diverse pathogens and nonpathogens.
|
|
Scooped by
Kamoun Lab @ TSL
August 27, 2012 7:16 PM
|
Compatible/incompatible interactions between the tomato wilt fungus Fusarium oxysporum f. sp. lycopersici (FOL) and tomato Solanum lycopersicum are controlled by three avirulence genes (AVR1–3) in FOL and the corresponding resistance genes (I–I3) in tomato. The three known races (1, 2 and 3) of FOL carry AVR genes in different combinations. The current model to explain the proposed order of mutations in AVR genes is: i) FOL race 2 emerged from race 1 by losing the AVR1 and thus avoiding host resistance mediated by I (the resistance gene corresponding to AVR1), and ii) race 3 emerged when race 2 sustained a point mutation in AVR2, allowing it to evade I2-mediated resistance of the host. Here, an alternative mechanism of mutation of AVR genes was determined by analyses of a race 3 isolate, KoChi-1, that we recovered from a Japanese tomato field in 2008. Although KoChi-1 is race 3, it has an AVR1 gene that is truncated by the transposon Hormin, which belongs to the hAT family. This provides evidence that mobile genetic elements may be one of the driving forces underlying race evolution. KoChi-1 transformants carrying a wild type AVR1 gene from race 1 lost pathogenicity to cultivars carrying I, showing that the truncated KoChi-1 avr1 is not functional. These results imply that KoChi-1 is a new race 3 biotype and propose an additional path for emergence of FOL races: Race 2 emerged from race 1 by transposon-insertion into AVR1, not by deletion of the AVR1 locus; then a point mutation in race 2 AVR2 resulted in emergence of race 3.
|
|
Scooped by
Kamoun Lab @ TSL
August 14, 2012 6:15 PM
|
In the beginning, there was the genome. Then came the foldome, the phenome and the connectome, quickly followed by the secretome, the otherome and the unknome. Over the past decade, a linguistic trickle swelled into a flood of buzzwords tagged with the curiously resonant suffix "ome." Today, hundreds of "omic" terms have worked their way into the lexicon, coined mostly by scientists intent on creating new sub-specialties. "It sounds futuristic. It sounds computational," said medical geneticist Robert C. Green at Harvard Medical School, who studies what he and his colleagues call the incidentalome—the realm of all incidental medical findings. "When you use the term "omics," it signals you are a new paradigm guy."
|
|
Scooped by
Kamoun Lab @ TSL
August 11, 2012 4:48 AM
|
Nicotiana benthamiana is a widely used model plant species for the study of fundamental questions in molecular plant-microbe interactions and other areas of plant biology. This popularity derives from its well-characterized susceptibility to diverse pathogens and especially its amenability to virus-induced gene silencing (VIGS) and transient protein expression methods. Here we report the generation of a 63-fold coverage draft genome sequence of N. benthamiana and its availability on the Sol Genomics Network (http://solgenomics.net/) for both BLAST searches and for downloading to local servers. The estimated genome size of N. benthamiana is ~3 gigabases (Gb). The current assembly consists of ~141,000 scaffolds, spanning 2.6 Gb of which >50% are longer than 89 kilobases. Of the ~16,000 N. benthamiana unigenes available in GenBank, >90% are represented in the assembly. The usefulness of the sequence was demonstrated by the retrieval of N. benthamiana orthologs for 24 immunity-associated genes from other species including Ago2, Ago7, Bak1, Bik1, Crt1, Fls2, Pto, Prf, Rar1 and MAP kinases. The sequence will also be useful for comparative genomics in the Solanaceae as shown here by the discovery of microsynteny between N. benthamiana and tomato in the region encompassing the Pto/Prf genes.
|
|
Scooped by
Kamoun Lab @ TSL
August 8, 2012 1:06 PM
|
FungiDB new release now includes genome sequence and gene expression data for six oomycetes: Phytophthora sojae, P. ramorum, P. infestans, P. parasitica, Hyaloperonospora arabidopsidis and Pythium ultimum, in addition to 25 fungal species. /via Brett Tyler
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
July 20, 2012 4:27 AM
|
This course is aimed at PhD students and post-doctoral researchers who are working on all aspects of fungal and oomycete-induced disease in plants. The primary aim of this course is to familiarize the participants with the toolset and data available for analysis of host and pathogen genomes, and infectious phenotypes. Lectures will provide an overview with the data available for plant pathogens, the technologies used to generate these data and the resources that make them available. Practicals will consist of computer exercises that will familliarise users with tools and resources at first hand, using relevent examples from the study of pathogenesis.
The course will cover primarily the PhytoPath resource, the associated Ensembl databases for plants, fungi and protists, and PHI-base. Additional tools and resources used in the analysis of genome-scale data will also be covered, including the Ondex software for network analysis. An introductory practical on genome assembly and alignment (using next-generation sequencing data) will also be provided.
|
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
December 7, 2012 8:40 AM
|
Horizontal gene transfer has been postulated to occur between crops to co-occurring parasitic plants, but empirical evidence has been lacking. We present evidence that an HGT event moved a nuclear monocot gene into the genome of the eudicot parasite witchweed (Striga hermonthica), which infects many grass species in Africa. Analysis of expressed sequence tags revealed that the genome of S. hermonthica contains a nuclear gene that is widely conserved among grass species but is not found in other eudicots. Phylogenetically, this gene clusters with sorghum genes, the monocot host of the parasitic weed, suggesting that nuclear genes can be captured by parasitic weeds in nature.
|
|
Scooped by
Kamoun Lab @ TSL
November 20, 2012 11:28 AM
|
Background - Oomycetes are fungal-like microorganisms evolutionary distinct from true fungi, belonging to the Stramenopile lineage and comprising major plant pathogens. Both oomycetes and fungi express proteins able to interact with cellulose, a major component of plant and oomycete cell walls, through the presence of carbohydrate-binding module belonging to the family 1 (CBM1). Fungal CBM1-containing proteins were implicated in cellulose degradation whereas in oomycetes, the Cellulose Binding Elicitor Lectin (CBEL), a well-characterized CBM1-protein from Phytophthora parasitica, was implicated in cell wall integrity, adhesion to cellulosic substrates and induction of plant immunity. Results - To extend our knowledge on CBM1-containing proteins in oomycetes, we have conducted a comprehensive analysis on 60 fungi and 7 oomycetes genomes leading to the identification of 518 CBM1-containing proteins. In plant-interacting microorganisms, the larger number of CBM1-protein coding genes is expressed by necrotroph and hemibiotrophic pathogens, whereas a strong reduction of these genes is observed in symbionts and biotrophs. In fungi, more than 70% of CBM1-containing proteins correspond to enzymatic proteins in which CBM1 is associated with a catalytic unit involved in cellulose degradation. In oomycetes more than 90% of proteins are similar to CBEL in which CBM1 is associated with a non-catalytic PAN/Apple domain, known to interact with specific carbohydrates or proteins. Distinct Stramenopile genomes like diatoms and brown algae are devoid of CBM1 coding genes. A CBM1-PAN/Apple association 3D structural modeling was built allowing the identification of amino acid residues interacting with cellulose and suggesting the putative interaction of the PAN/Apple domain with another type of glucan. By Surface Plasmon Resonance experiments, we showed that CBEL binds to glycoproteins through galactose or N-acetyl-galactosamine motifs. Conclusions - This study provides insight into the evolution and biological roles of CBM1-containing proteins from oomycetes. We show that while CBM1s from fungi and oomycetes are similar, they team up with different protein domains, either in proteins implicated in the degradation of plant cell wall components in the case of fungi or in proteins involved in adhesion to polysaccharidic substrates in the case of oomycetes. This work highlighted the unique role and evolution of CBM1 proteins in oomycete among the Stramenopile lineage.
|
|
Scooped by
Kamoun Lab @ TSL
October 9, 2012 4:42 AM
|
The button mushroom occupies a prominent place in our diet and in the grocery store where it boasts a tasty multibillion-dollar niche, while in nature, Agaricus bisporus is known to decay leaf matter on the forest floor. Now, owing to an international collaboration of two-dozen institutions led by the French National Institute for Agricultural Research (INRA) and the U.S. Department of Energy Joint Genome Institute (DOE JGI), the full repertoire of A. bisporus genes has been determined. In particular, new work shows how its genes are actually deployed not only in leaf decay but also wood decay and in the development of fruiting bodies (the above ground part of the mushroom harvested for food). The work also suggests how such processes have major implications for forest carbon management. The analysis of the inner workings of the world’s most cultivated mushroom was published online the week of October 8 in the journal, the Proceedings of the National Academy of Sciences (PNAS).
|
|
Scooped by
Kamoun Lab @ TSL
October 5, 2012 9:33 AM
|
|
|
Scooped by
Kamoun Lab @ TSL
October 2, 2012 4:09 AM
|
Plants and pathogens evolve in response to each other. This co-evolutionary arms race is fueled by genetic variation underlying the recognition of pathogen proteins by the host and the defeat of host defenses by the pathogen. Together with new mutations, genetic diversity in populations of both the host and pathogen represent a pool of possible variants to maintain adaptation via natural selection.Drastic changes in genetic diversity in crop species have occurred as a consequence of domestication. Whether changes in the genetic composition of these host populations also have affected genetic diversity in pathogen species is, so far, poorly understood. Advances in comparative genomics and population genomic approaches open new avenues to study adaptive processes in plant pathogens and to infer the impact of agro-ecosystems on the evolution of pathogen populations. Here we summarize new insights gained from comparative genome studies and population genomics in host-pathogen systems.
Via Nicolas Denancé
|
|
Scooped by
Kamoun Lab @ TSL
September 6, 2012 10:34 AM
|
Biotroph pathogens establish intimate interactions with their hosts that are conditioned by the successful secretion of effectors in infected tissues and subsequent manipulation of host physiology. The identification of early-expressed pathogen effectors and early-modulated host functions is currently a major goal to understand the molecular basis of biotrophy. Here, we report the 454-pyrosequencing transcriptome analysis of early stages of poplar leaf colonization by the rust fungus Melampsora larici-populina. Among the 841,301 reads considered for analysis, 616,879 and 649 were successfully mapped to Populus trichocarpa and M. larici-populina genome sequences, respectively. From a methodological aspect, these results indicate that this single approach is not appropriate to saturate poplar transcriptome and to follow transcript accumulation of the pathogen. We identified 19 pathogen transcripts encoding early-expressed small-secreted proteins representing candidate effectors of interest for forthcoming studies. Poplar RNA-Seq data were validated by oligoarrays and quantitatively analysed, which revealed a highly stable transcriptome with a single transcript encoding a sulfate transporter (herein named PtSultr3;5, POPTR_0006s16150) showing a dramatic increase upon colonization by either virulent or avirulent M. larici-populina strains. Perspectives connecting host sulfate transport and biotrophic lifestyle are discussed.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
August 28, 2012 7:25 AM
|
• Stagonospora nodorum: From Pathology to Genomics and Host Resistance Richard P. Oliver, Timothy L. Friesen, Justin D. Faris, and Peter S. Solomon • Pathogenomics of the Ralstonia solanacearum Species Complex Stéphane Genin and Timothy P. Denny • The Genomics of Obligate (and Nonobligate) Biotrophs Pietro D. Spanu • Genome-Enabled Perspectives on the Composition, Evolution, and Expression of Virulence Determinants in Bacterial Plant Pathogens Magdalen Lindeberg • Plant Defense Compounds: Systems Approaches to Metabolic Analysis Daniel J. Kliebenstein • Role of Nematode Peptides and Other Small Molecules in Plant Parasitism Melissa G. Mitchum, Xiaohong Wang, Jianying Wang, and Eric L. Davis • Somatic Hybridization in the Uredinales Robert F. Park and Colin R. Wellings • Plant Immunity to Necrotrophs Tesfaye Mengiste • Mechanisms and Evolution of Virulence in Oomycetes Rays H.Y. Jiang and Brett M. Tyler • Variation and Selection of Quantitative Traits in Plant Pathogens Christian Lannou • Gall Midges (Hessian Flies) as Plant Pathogens Jeff J. Stuart, Ming-Shun Chen, Richard Shukle, and Marion O. Harris • Phytophthora Beyond Agriculture Everett M. Hansen, Paul W. Reeser, and Wendy Sutton • Diversity and Natural Functions of Antibiotics Produced by Beneficial and Plant Pathogenic Bacteria Jos M. Raaijmakers and Mark Mazzola • The Role of Secretion Systems and Small Molecules in Soft-Rot Enterobacteriaceae Pathogenicity Amy Charkowski, et al. • Receptor Kinase Signaling Pathways in Plant-Microbe Interactions Meritxell Antolín-Llovera, Martina K. Ried, Andreas Binder, and Martin Parniske • Fire Blight: Applied Genomic Insights of the Pathogen and Host Mickael Malnoy, Stefan Martens, John L. Norelli, Marie-Anne Barny, George W. Sundin, Theo H.M. Smits, and Brion Duffy
|
|
Scooped by
Kamoun Lab @ TSL
August 14, 2012 6:29 PM
|
|
|
Scooped by
Kamoun Lab @ TSL
August 13, 2012 4:53 PM
|
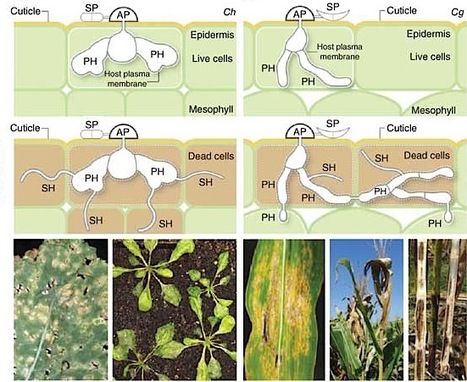
Colletotrichum species are fungal pathogens that devastate crop plants worldwide. Host infection involves the differentiation of specialized cell types that are associated with penetration, growth inside living host cells (biotrophy) and tissue destruction (necrotrophy). We report here genome and transcriptome analyses of Colletotrichum higginsianum infecting Arabidopsis thaliana and Colletotrichum graminicola infecting maize. Comparative genomics showed that both fungi have large sets of pathogenicity-related genes, but families of genes encoding secreted effectors, pectin-degrading enzymes, secondary metabolism enzymes, transporters and peptidases are expanded in C. higginsianum. Genome-wide expression profiling revealed that these genes are transcribed in successive waves that are linked to pathogenic transitions: effectors and secondary metabolism enzymes are induced before penetration and during biotrophy, whereas most hydrolases and transporters are upregulated later, at the switch to necrotrophy. Our findings show that preinvasion perception of plant-derived signals substantially reprograms fungal gene expression and indicate previously unknown functions for particular fungal cell types.
|
|
Scooped by
Kamoun Lab @ TSL
August 9, 2012 1:31 AM
|
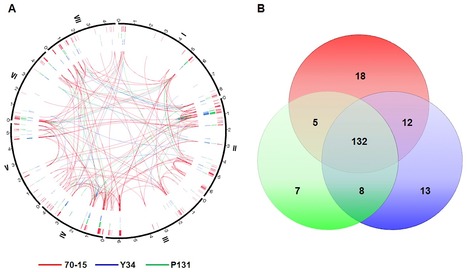
Rice blast caused by Magnaporthe oryzae is one of the most destructive diseases of rice worldwide. The fungal pathogen is notorious for its ability to overcome host resistance. To better understand its genetic variation in nature, we sequenced the genomes of two field isolates, Y34 and P131. In comparison with the previously sequenced laboratory strain 70-15, both field isolates had a similar genome size but slightly more genes. Sequences from the field isolates were used to improve genome assembly and gene prediction of 70-15. Although the overall genome structure is similar, a number of gene families that are likely involved in plant-fungal interactions are expanded in the field isolates. Genome-wide analysis on asynonymous to synonymous nucleotide substitution rates revealed that many infection-related genes underwent diversifying selection. The field isolates also have hundreds of isolate-specific genes and a number of isolate-specific gene duplication events. Functional characterization of randomly selected isolate-specific genes revealed that they play diverse roles, some of which affect virulence. Furthermore, each genome contains thousands of loci of transposon-like elements, but less than 30% of them are conserved among different isolates, suggesting active transposition events in M. oryzae. A total of approximately 200 genes were disrupted in these three strains by transposable elements. Interestingly, transposon-like elements tend to be associated with isolate-specific or duplicated sequences. Overall, our results indicate that gain or loss of unique genes, DNA duplication, gene family expansion, and frequent translocation of transposon-like elements are important factors in genome variation of the rice blast fungus.
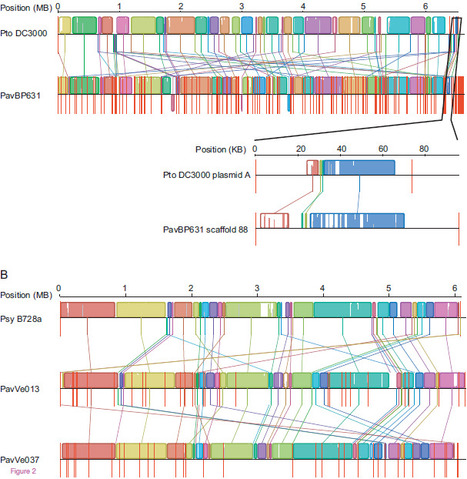
Hazelnut (Corylus avellana) decline disease in Greece and Italy is caused by the convergent evolution of two distantly related lineages of Pseudomonas syringae pv. avellanae (Pav). We sequenced the genomes of three Pav isolates to determine if their convergent virulence phenotype had a common genetic basis due to either genetic exchange between lineages or parallel evolution. We found little evidence for horizontal transfer (recombination) of genes between Pav lineages, but two large genomic islands (GIs) have been recently acquired by one of the lineages. Evolutionary analyses of the genes encoding type III secreted effectors (T3SEs) that are translocated into host cells and are important for both suppressing and eliciting defense responses show that the two Pav lineages have dramatically different T3SE profiles, with only two shared putatively functional T3SEs. One Pav lineage has undergone unprecedented secretome remodeling, including the acquisition of eleven new T3SEs and the loss or pseudogenization of 15, including five of the six core T3SE families that are present in the other Pav lineage. Molecular dating indicates that divergence within both of the Pav lineages predates their observation in the field. This suggest that both Pav lineages have been cryptically infecting hazelnut trees or wild relatives for many years, and that the emergence of hazelnut decline in the 1970s may have been due to changes in agricultural practice. These data show that divergent lineages of P. syringae can converge on identical disease etiology on the same host plant using different virulence mechanisms and that dramatic shifts in the arsenal of T3SEs can accompany disease emergence.
Via Nicolas Denancé
|