Your new post is loading...
Your new post is loading...

|
Scooped by
Jean-Michel Ané
Today, 12:46 PM
|
The specific partnership between legumes and rhizobia relies on a chemical dialogue. Plant flavonoids activate the bacterial transcription factor NodD, which triggers production of Nod factors that are recognized by the plant. Structural studies of the Pisum sativum (pea) symbiont Rhizobium leguminosarum NodD revealed two pockets that are essential for its activation by flavonoids. Comparative studies with NodD1 of Sinorhizobium medicae, the symbiont of Medicago truncatula, revealed that this specificity is determined by the shape of the pocket and by specific amino acids. A chimeric NodD containing the flavonoid recognition residues from S. medicae NodD1 in the R. leguminosarum NodD backbone was sufficient to complement nitrogen fixation in M. truncatula by an S. medicae nodD1 mutant, confirming the critical role of flavonoid recognition in host range.
|
|
Scooped by
Jean-Michel Ané
January 9, 7:30 PM
|
During host-microbe symbioses, the fitness of mutualistic microbes is determined by the interactions that concurrently occur, throughout their life cycle, with their host and other members of the surrounding microbial community. Disentangling how these multiple interactions shape the fitness of microbial symbionts is challenging, but is essential to understand the diversity and functioning of mutualisms. Here we examined the different fitness components of rhizobial symbionts of the legume plant Mimosa pudica across the multiple stages of their symbiotic life cycle. By comparing rhizobial symbiotic fitness in single and pairwise inoculations, we found that inter-bacterial interactions causing significant fitness effects are common, transitive and can have major consequences, sometimes leading to the extinction of a strain. These interactions predominantly occur at the root infection (nodulation) step, but smaller post-infection interaction effects, involving yet uncharacterized mechanisms, were also detected. Furthermore, considering pairwise interactions was sufficient to predict fitness ranks in more complex rhizobial communities consisting of 6 or 8 strains, indicating that higher-order interaction effects do not play a significant role in these communities. Overall, our results provide a quantitative framework to describe the main drivers of rhizobial symbiotic fitness in a simple community context.
|
|
Scooped by
Jean-Michel Ané
January 8, 11:38 AM
|
The Definitive Handbook of Azospirillum" stands out for its comprehensive and specialized focus on Azospirillum, its detailed analysis of the evolutionary knowledge, and its practical experimental proposals. It offers a detailed examination of Azospirillum, while other texts tend to provide broader, more general, or technical perspectives on related topics.
This book not only explores the biological and ecological significance of Azospirillum but also covers the various roles it plays in different environments, especially in agriculture where it has potential applications as a biofertilizer. By providing both historical context and cutting-edge research, this book bridges the gap between past discoveries and future possibilities, making it a valuable tool for researchers and professionals.
|
|
Scooped by
Jean-Michel Ané
January 5, 3:48 PM
|
The root nodule symbiosis of plants with nitrogen-fixing bacteria is phylogenetically restricted to a single clade of flowering plants, which calls for as yet unidentified trait acquisitions and genetic changes in the last common ancestor. Here we discovered—within the promoter of the transcription factor gene Nodule Inception (NIN)—a cis-regulatory element (PACE), exclusively present in members of this clade. PACE was essential for restoring infection threads in nin mutants of the legume Lotus japonicus. PACE sequence variants from root nodule symbiosis-competent species appeared functionally equivalent. Evolutionary loss or mutation of PACE is associated with loss of this symbiosis. During the early stages of nodule development, PACE dictates gene expression in a spatially restricted domain containing cortical cells carrying infection threads. Consistent with its expression domain, PACE-driven NIN expression restored the formation of cortical infection threads, also when engineered into the NIN promoter of tomato. Our data pinpoint PACE as a key evolutionary invention that connected NIN to a pre-existing symbiosis signal transduction cascade that governs the intracellular accommodation of arbuscular mycorrhiza fungi and is conserved throughout land plants. This connection enabled bacterial uptake into plant cells via intracellular support structures such as infection threads, a unique and unifying feature of this symbiosis.
|
|
Scooped by
Jean-Michel Ané
January 2, 4:43 PM
|
Alfalfa (Medicago sativa L.), known as “Queen of forages”, is valued to its high-nutritional quality and is a key member of Leguminosae family. Its productivity is largely attributed to mutualistic symbioses with arbuscular mycorrhizal fungi (AMF) and rhizobia, which facilitate nutrient exchange and plant growth. However, the coexistence and mutualistic interactions between rhizobia and AMF across alfalfa genotypes with differing yields in native soil remain poorly understood. In this study, we investigated the community composition of rhizobia and AMF colonizing alfalfa roots across different-yield varieties. Our results showed variations in dominant microbial taxa and the structural complexity of root-associated microbial networks among genotypes. Moreover, rhizobia exhibited no significant associations with AMF on genus level, however, negative correlations were observed among genera within the AMF community, and a comparable trend was identified among rhizobial taxa. In summary, our findings offer new insights into how native soil microbiota influence the dual symbiotic relationships of alfalfa, with implications for leveraging native microbial communities to enhance sustainable forage production.
|
|
Scooped by
Jean-Michel Ané
December 31, 2025 4:13 PM
|
Arbuscular mycorrhizal (AM) associations of plants and Glomeromycotina soil fungi play a crucial role in all terrestrial ecosystems. In this mutually beneficial interaction, obligate biotrophic fungi acquire photosynthetically fixed carbon from the plant, while the mutualistic fungi enhance plant access to soil nutrients. AM fungi colonize the inner tissues of host roots, where they form specialized symbiotic structures (arbuscules) within fully differentiated cortex cells that are reprogrammed to host the microbe. Given the intimate nature of the interaction, extensive partner communication at the interface of plant and fungal cells is crucial for the development and functioning of AM symbiosis. The peri-arbuscular space, a specialized apoplast compartment surrounding the arbuscules, supports not only nutrient exchange between the symbiotic partners but is also the site of extensive partner crosstalk mediated by cell wall components, receptors, signaling peptides, and extracellular vesicles. Such signaling processes in the apoplast modulate plant immune responses to enable colonization by beneficial fungi, making this compartment a key player for the establishment and maintenance of AM symbiosis. In this review, we discuss recent discoveries related to the role of partner communication in the apoplast, with a focus on peptide and cell wall signaling, as well as extracellular vesicles.
|
|
Scooped by
Jean-Michel Ané
December 31, 2025 4:10 PM
|
Ectomycorrhizal fungi form symbiotic relationships with a wide range of terrestrial plants, acquiring carbohydrates for themselves and promoting nutrient uptake in their host plants. However, some ectomycorrhizal fungi cannot effectively obtain the thiamine necessary for growth from their host or synthesize it themselves. Ectomycorrhizal fungi can recruit hypha-associated microorganisms, which play a vital role in promoting nutrient absorption and ectomycorrhizal root formation, ultimately colonizing within fruiting bodies to form a unique bacterial microbiota. In this study, non-targeted metabolomics and whole-genome sequencing were employed to investigate the colonization characteristics of the hyphae-associated bacterium Bacillus altitudinis B4 on the mycelial surface of ectomycorrhizal fungus Suillus clintonianus, as well as the synergistic promotion of thiamine synthesis and absorption by B. altitudinis B4 and the fungal mycelium, respectively. The results suggested that S. clintonianus first secreted ureidosuccinic acid and pregnenolone, recruiting the hyphae-associated bacterium B. altitudinis B4 to the mycelial surface. Subsequently, the ureidosuccinic acid secreted by S. clintonianus further stimulated B. altitudinis B4 to enhance thiamine production by increasing its biomass and upregulating the expression of related functional genes. Finally, S. clintonianus absorbed the thiamine secreted by the B. altitudinis B4, promoting fungal growth and increasing the colonization rate in association with Pinus massoniana. This study elucidates the thiamine acquisition mechanisms of ectomycorrhizal fungi, highlighting the critical role of bacterial partners in fungal nutrition and host-fungal interactions.
|
|
Scooped by
Jean-Michel Ané
December 31, 2025 3:43 PM
|
In the current context of climate change, there is a need to develop more sustainable agrifood strategies. As an alternative to the intensive use of chemically synthesized fertilizers and pesticides that pollute water and impact biodiversity, there is a growing interest in using beneficial microbes as biostimulants and/or bioprotection agents. However, their implementation in agriculture remains a challenge due to highly variable outcomes and benefits. Furthermore, there are major knowledge gaps about the molecular mechanisms that regulate different plant–microbe interactions. In the present review, we summarize current knowledge on the molecular mechanisms that control different beneficial plant root–microbe interactions; namely, arbuscular mycorrhiza, the rhizobium–legume symbiosis, ectomycorrhiza, and fungal and bacterial endophytic associations. This includes the signaling pathways required for recognition of microbes as beneficial, the metabolic pathways that provide nutritional benefits to the plant, and the regulatory pathways that modulate the extent of symbiosis establishment depending on soil nutrient availability and plant needs. Our aim is to highlight the main common mechanisms, as well as knowledge gaps, in order to promote the use of microbes, either individually or in consortia, within the framework of a sustainable agriculture that is less dependent on chemicals and more protective of biodiversity and water resources.
|
|
Scooped by
Jean-Michel Ané
December 28, 2025 4:57 PM
|
PerCon SFA project data dentification of spatially resolved biomarkers of drought in Sorghum bicolor rhizosphere molecular-microbe interactions using a novel root cartography "RhizoGrid" system for sampling plants under drought and control conditions across 10 equally sized root zone environments (4 quadrants each). Each quadrant was sampled and processed for 16S amplicon, metabolomics, and X-ray computed tomography (XCT). Data download includes experimental metadata and results files for 16S rRNA sequence analysis of microbial community assembly (processed data files), liquid chromatography mass spectrometry (LC-MS) metabolomics analysis of microbial community root exudates (processed data files), X-ray computed tomography (XCT) spatial gradient analysis (raw and processed data files) of microbial community composition, and related computational modeling outputs.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 9:34 PM
|
Background and aims
Mycorrhizal (AMF) benefits to ancient tetraploid and hexaploid wheats, particularly under saline conditions, are not sufficiently known.
Methods
A two-year field experiment and a pot experiment were carried out, where the field experiment encompassed non-saline and saline (120 mM NaCl) irrigation, presence and absence of AMF (Funneliformis mosseae) inoculation, and 10 wheat genotypes. The pot experiment encompassed four salinities (0, 40, 80, and 120 mM) and two levels of AMF inoculation (with and without of AMF inoculation) and 11 genotypes.
Results
Salinity suppressed the chlorophyll, carotenoids, K, and P, grains/m2, grain yield, harvest index, and dry mass. Though, it boosted the activities of antioxidative enzymes, Na, electrolyte leakage, Na/K, protein, wet gluten, and gluten index. Inoculation to AMF led to enhancement in the maximum quantum efficiency of photosystem II, chlorophyll, K, P, N, and total phenolic compounds concentrations, the activities of antioxidative enzymes, grains/m2, grain yield, dry mass, protein, wet gluten, and gluten index, while decreasing the Na concentration, Na/K, and electrolyte leakage, particularly in the salt-stricken plants; favorable responses to the AMF were more appreciable in the salt-stricken modern wheats, than the ancient emmer and spelt wheats.
Conclusion
Salinity and AMF exerted contrasting effects on physiological, growth, dry mass, and grain yield attributes of different genotypes, with a tendency of salt-induced suppressions and AMF-induced enhancements to be less notable in the ancient wheats, than the modern bread and durum wheats. Though, salinity and AMF inoculation shared a same trend in improving the grain and flour quality attributes.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 9:24 PM
|
This thesis tests the hypothesis that phosphorus (P) fertiliser addition can optimise nitrogen (N2) fixation processes and increase soybean yield. Using a combination of controlled environment studies, field trial and global meta-analysis, this thesis aids in achieving sustainable soybean production through building improved understanding of the effects of P fertiliser addition to inform future fertiliser guidelines, crop models and management practices. Global meta-analysis showed an increase in soybean response to P fertiliser addition with seed yield increasing by 25%. This also highlighted the complexity of soybean yield response to P fertiliser, with a several key management and environmental conditions having a significant effect, including soil P concentration, pH, fertiliser type and rate of application and climatic conditions – indicating soybean yield cannot be increased by single P fertiliser applications alone. Controlled environment studies revealed P addition significantly increased nitrogen (N2) fixation. Key nodule traits significantly correlated with shoot N; however, further work should examine the mechanistic pathways driving the increase in nodule formation. Interestingly, controlled environment studies revealed nodule function was not influenced by P fertiliser addition. Instead, regulatory mechanisms such as maintenance of nodule P concentration and leghaemoglobin concentration under low P conditions maintained N2 fixation. Through combined analysis of multiple growth parameters and measures of plant physiology, seed yield was found to increase under P fertiliser addition. Seed P concentration also increased following P fertiliser addition. Results of this thesis contribute to our understanding of soybean response to P fertiliser addition, particularly the improvement to key nodule traits to improve N2 fixation and the partitioning and remobilisation of resources to improve yield. This now needs to be upscaled at differing environmental and management conditions and incorporated into crop models to ensure the sustainable use of P fertiliser in soybean production globally.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 7:27 PM
|
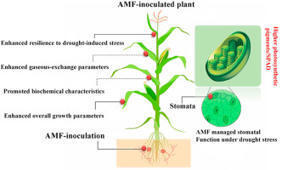
Drought-induced stress is a significant constraint for crop yields in semi-arid and arid areas.
Yield assessments under water stress indicate that mycorrhizae can alleviate the detrimental impacts of drought, placing them as sustainable options for agricultural practices in affected areas. Thus, we executed a two-year study to examine the effects of root colonization by two AMF species (Diversispora epigaea and Diversispora versiformis) under different drought stress conditions, assessing maize morpho-physiological and biochemical characteristics, nutrient absorption, yield components, oil percentage, and irrigation water efficiency. The research was conducted in a desolate region of Pakistan during the 2023 and 2024 growing seasons. Drought-induced stress was generated at two levels by irrigating after 80 % and 60 % water loss, categorized as severe and mild drought stress. Irrigation after a 40 % reduction in water was considered normal (without stress). The findings demonstrated that regardless of AMF species and level of drought stress, inoculated plants yielded heavier seeds, higher dry matter, chlorophyll (37 %) and carotenoids (41 %), phytohormone (27 %), enhanced oil yields (32 %) and seeds (24.2 %) compared to uninoculated plants. Notably, the maize seed yields of Diversispora epigaea-treated plants under every irrigation treatment surpassed those of Diversispora versiformis inoculated plants and uninoculated plants. Drought stress reduced nitrogen levels in seeds and leaves, whereas AMF enhanced nitrogen levels, particularly when crops were treated with Diversispora epigaea. Moreover, seed phosphorus percentages were not influenced by AMF in 2023. Conversely, the highest phosphorus percentages in seeds and leaves were recorded in crops inoculated with Diversispora epigaea in 2023. Our findings indicate that Diversispora epigaea exhibits greater efficiency under water stress and provides superior support to maize plants.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 7:17 PM
|
•Glomalin is widely used as a soil health and mycorrhizal fungi indicator.
•The extraction method raises concerns about its specificity to mycorrhizal fungi.
•Glomalin content correlates with plant litter decomposition and added organic matter.
•High-temperature citrate extracts include diverse compounds, not just glomalin.
•Glomalin is unsuitable for standardized measurements of mycorrhizal fungi.
|
|
|
Scooped by
Jean-Michel Ané
January 10, 7:17 PM
|
Background
Since the first description of an Azospirillum-like bacterium in 1925 by Martinus Beijerinck in The Netherlands, this genus has become a cornerstone of plant–microbe interactions and sustainable agriculture worldwide. Over the past century, Azospirillum has been extensively studied for its ability to promote plant growth, enhance stress tolerance, and contribute to nutrient acquisition, particularly in cereals and legumes. These functions are mediated by multiple mechanisms, including nitrogen fixation, phytohormone production, and the modulation of the root architecture by effector molecules.
Scope
This review presents a comprehensive synthesis of the historical and current research on Azospirillum and its impact in agriculture and beyond. It explores its taxonomic expansion, physiological versatility, genetic manipulation, and interactions with plant hosts and other microorganisms. It also examines its agronomical impact on extensive and intensive cropping systems, both individually and mixed in microbial consortia. Advances in formulation technologies and regulatory frameworks for commercial inoculants are discussed, as well as cutting-edge tools such as artificial intelligence and multi-omics integration that are reshaping how we understand and deploy this bacterium. Beyond agriculture, Azospirillum has proven valuable in environmental contexts such as revegetation of degraded lands, bioremediation of contaminated soils, and ecological restoration in arid zones. Its capacity to colonize diverse hosts, survive extreme conditions, and contribute to ecosystem processes underscores its potential far beyond agriculture.
Conclusion
One hundred years after its first scientific mention, Azospirillum remains not only relevant but also vital. As the world moves toward more sustainable agricultural and ecological systems, this review reaffirms the legacy and promise of this genus.
|
|
Scooped by
Jean-Michel Ané
January 8, 2:06 PM
|
Aspergillus fumigatus is a notorious pathogenic fungus responsible for various harmful, sometimes lethal, diseases known as aspergilloses. Understanding the gene regulatory networks that specify the expression programs underlying this fungus’ diverse phenotypes can shed mechanistic insight into its growth, development, and determinants of pathogenicity. We used eighteen publicly available RNA-seq datasets of Aspergillus fumigatus to construct a comprehensive gene regulatory network resource. Our resource, named GRAsp (Gene Regulation of Aspergillus fumigatus), was able to recapitulate known regulatory pathways such as response to hypoxia, iron and zinc homeostasis, and secondary metabolite synthesis. Further, GRAsp was experimentally validated in two cases: one in which GRAsp accurately identified an uncharacterized transcription factor negatively regulating the production of the virulence factor gliotoxin and another where GRAsp revealed the bZip protein, AtfA, as required for fungal responses to microbial signals known as lipo-chitooligosaccharides. Our work showcases the strength of using network-based approaches to generate new hypotheses about regulatory relationships in Aspergillus fumigatus. We also unveil an online, user-friendly version of GRAsp available to the Aspergillus research community.
|
|
Scooped by
Jean-Michel Ané
January 5, 9:24 PM
|
Rhizobial technology has become a transformative tool for environmentally friendly and sustainable agriculture. Rhizobia are key nitrogen-fixing bacteria that enhance soil fertility and reduce reliance on synthetic nitrogen fertilisers. In addition to nitrogen fixation, they act as effective plant growth promoters by producing phytohormones, mobilising nutrients, and improving root development. Advances in bioinoculant engineering now support efficient symbiotic associations in both leguminous and non-leguminous crops, offering a green strategy to boost agricultural productivity. Rhizobia also help plants withstand abiotic and biotic stresses, and many strains display strong biocontrol abilities by producing antimicrobial compounds and suppressing phytopathogens. However, their field performance can be inconsistent due to poor survival during storage, competition with native microbes, environmental conditions, and limited farmer awareness. To overcome these challenges, strategies such as co-inoculation with compatible microbes, encapsulated formulations, genetic enhancement, improved agronomic practices, pathogen management, and farmer awareness are being developed to increase inoculant stability and effectiveness. Overall, rhizobial technology serves as a cornerstone of smart, sustainable farming, supporting food security, environmental protection, and the restoration of soil health for future green agriculture.
|
|
Scooped by
Jean-Michel Ané
January 4, 8:50 PM
|
Modern agriculture faces an urgent need to improve nutrient use efficiency while reducing environmental impacts. Here, we show that ancestral traits controlling rhizosphere microbiome functions can be reintroduced into elite maize through targeted teosinte introgressions. Using near-isogenic lines, we mapped microbiome-associated phenotypes (MAPs) derived from teosinte that suppress nitrification and denitrification—key microbial processes contributing to nitrogen loss. These introgressions altered root exudate chemistry, resulting in distinct microbial assemblies and enhanced nitrogen retention. We identified candidate loci and exudate metabolites responsible for suppressive activity and demonstrated their functional effects in vitro. These findings reveal a genetic and biochemical basis for rewilding microbiome-mediated ecosystem services in crops, offering a scalable path toward sustainable nutrient management in global agriculture.
|
|
Scooped by
Jean-Michel Ané
January 2, 4:41 PM
|
Cytokinin (CK) is a key regulator of root system architecture including primary root (PR) and lateral root (LR) development. During root nodule symbiosis in legumes, CK promotes nodule organogenesis in the root cortex while simultaneously suppressing infection thread (IT) formation in the epidermis, yet the molecular mechanisms enabling this spatial specificity remain incompletely understood. TCP INTERACTOR CONTAINING EAR MOTIF PROTEIN (TIE) proteins negatively regulate CK signaling in Arabidopsis roots, but whether legume orthologs modulate symbiotic CK responses remains unknown.
We characterized Lotus tie1 loss-of-function mutants through root and nodulation assays, transcriptomics, confocal microscopy of CK signaling reporter TCSn, and ethylene quantification.
LjTIE1 expression was induced by CK and Nod-factor, tie1 mutants exhibited reduced PR length, LR density, IT formation, and nodule number alongside elevated ethylene emission - phenotypes fully rescued by ethylene biosynthesis inhibition. Despite enhanced CK signaling (confirmed via TCSn reporter and transcriptomics), tie1 roots formed no spontaneous nodules but displayed hypernodulation under exogenous CK treatment.
LjTIE1 provides temporal control of symbiosis by dampening CK signaling after nodule initiation, revealing a regulatory layer that prevents constitutive nodulation despite elevated CK perception.
|
|
Scooped by
Jean-Michel Ané
December 31, 2025 4:11 PM
|
Soil, the Earth's upper crust layer, is crucial for ecological processes, comprising mineral, organic, and biological components that determine fertility and multifuncionality. Human-induced degradation necessitates advancements in pedology and soil conservation. The rhizosphere, surrounding plant roots, houses a diverse microbial community, notably bacteria, which enhance plant growth and disease resistance. Root exudates fuel biological activity and nutrient cycling, supporting microbial growth, improving soil structure, and reducing plant stress. Plant-microorganism interactions in ecological and agricultural systems play a vital role for maintaining primary production and ecosystem sustainability. Moreover, arbuscular mycorrhizae and nitrogen-fixing bacteria are essential, influencing plant development, sustainability, and ecosystem health. Specific bacterial phyla populate the rhizosphere and endosphere, with Plant Growth-Promoting Rhizobacteria (PGPR), such as Pseudomonas spp. and Bacillus spp., playing a prominent role. PGPR employ direct and indirect mechanisms, including phytohormone production, mineral solubilization, systemic resistance induction, antibiosis, competition for resources, and ACC deaminase activity, The amalgamation of these traits underscores the conceptual foundation for comprehending the ecological and agricultural implications of employing microbes. This inquiry is particularly relevant to sustainable agriculture, where the use of microbes, including PGPR, plays a crucial role in biofertilization and mitigating environmental stressors. Thus, investigating the ecological and agricultural implications through multi-omics approaches such as genomics, transcriptomics, proteomics, and metabolomics offers valuable insights. The integration of these multi-omics data provides a comprehensive framework for understanding the complex interactions between plants, bacteria, and fungi. This holistic perspective not only deepens our understanding of soil ecology but also lays the groundwork for informed and sustainable agricultural practices, fostering resilience against environmental stresses.
|
|
Scooped by
Jean-Michel Ané
December 31, 2025 3:50 PM
|
Microbiota-mediated nutrient turnover in the rhizosphere determines nutrient bioavailability, thereby enhancing nutrient uptake, utilization, and ultimately crop productivity. Consequently, elucidating the functional core microbiota in rhizosphere nutrient turnover is of critical importance. In this study, we leveraged soybean germplasm core collections to investigate the tripartite relationship among host genotype, core microbiota and nutrient availability, with a focus on delineating the pivotal role of core microbiota in nutrient turnover. Our results suggest that phylogenetic variation significantly shape root-associated microbial communities and rhizosphere nutrient availability, explaining 11.75 % and 2.07 % of total variances, respectively. Core microbiota analysis identified 29 phylogenetic conserved core amplicon sequence variants (ASVs), the majority of which exhibited significant correlated with nutrient availability. Notably, three key core ASVs—ASV13, ASV14 and ASV12, positively correlated with alkali-hydrolyzed nitrogen, available phosphorus, and soil organic matter, respectively. These taxa were subsequently incorporated into a Bradyrhizobium-based synthetic bacterial community (SynCom) to validate their functional roles. Further experiments confirmed that core microbiota-driven nutrient turnover directly facilitates host plant, as evidenced by SynCom inoculation assays. Collectively, this study establishes that phylogenetically conserved core microbiota critically regulate nutrient turnover and acquisition efficiency in the rhizosphere. These insights advance our understanding the ecological function of core microbiota in the rhizosphere and provide a framework for harnessing the beneficial traits in sustainable agriculture.
|
|
Scooped by
Jean-Michel Ané
December 28, 2025 5:02 PM
|
Transmission electron microscopy was the key for revealing structural similarities between intracellular plant-microbe interactions.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 9:43 PM
|
Arbuscular mycorrhizal fungi (AMF) are obligate biotrophs that rely on host-derived symbiotic carbohydrates. However, it remains unclear whether symbiotic AMF can access exogenous non-symbiotic carbon sources, complicating our understanding of their relationship with host plants. Here, we investigated the direct uptake of exogenous 13C1-labeled myristate by three symbiotic AMF species (Rhizophagus irregularis, R. intraradices, and R. diaphanous) and assessed their growth responses using AMF-carrot hairy root co-culture systems. Furthermore, we explored the environmental distribution of myristate, and evaluated the impact of exogenous myristate on the carbon-phosphorus exchange between R. irregularis and alfalfa or rice in a greenhouse experiment. Symbiotic AMF can absorb exogenous myristate, as evidenced by 13C enrichment and transcriptional activation of fatty acid transport and metabolism genes in AMF extraradical hyphae. Myristate is commonly present in various soil and plant environments, and its application increased both intraradical and extraradical fungal biomass, possibly linked to suppressed mycorrhizal-activated defense responses in host roots. Unexpectedly, exogenous myristate reduced the mycorrhizal phosphorus benefits for both alfalfa and rice and decreased their symbiotic carbon allocation to root-colonizing AMF, although these effects varied with soil phosphorus conditions. These findings provide new insights into understanding and manipulating the nutritional interactions between AMF and host plants.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 9:27 PM
|
Crop rotation enhances agroecosystem sustainability by reducing nutrient loss, improving soil fertility, and decreasing crop evapotranspiration. Arbuscular mycorrhizal fungi (AMF) inoculation further supports enhanced nutrient and water uptake by plants, potentially improving water use efficiency and soil health while reducing fertigation needs. However, crop species may respond differently to AMF inoculation under varying fertigation regimes. In this study, the response of four horticultural species to AMF inoculation was investigated under optimal (100 %) and deficit (25 % reduction) water and fertilizer (fertigation) availability. Leek, courgette, white bean, and celery were planted consecutively over the course of two years, with crop production and quality as well as leaf nutrients and soil parameters being measured at the end of each crop cycle. Mycorrhizal inoculation did not improve any agronomic parameter among the crops studied under either fertigation condition.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 9:16 PM
|
The symbiotic nitrogen fixation process between legume roots and rhizobia initiates at the root hairs. Rhizobia initially colonize the tip of the root hairs and induce its curling to become entrapped. However, the specific molecular mechanisms underlying root hair deformation and curling in response to rhizobial infection remain unclear. In this study, transcriptome analysis of wild-type JiMa389 and nodulation-deficient mutant jima61 of Melilotus albus, the Rho-like small GTPase MaROP10. Our results show that MaROP10 functions as an interacts with the Nod factor receptor NFR5 to regulate rhizobia-induced root hair deformation and infection thread formation during the early stages of rhizobial infection. To elucidate the mechanism of MaROP10, we further identified its downstream potential effector protein, MaRIC6, which positively regulates root hair deformation and infection thread formation. Taken together, MaROP10 likely integrates signals from the symbiotic receptor NFR5 to regulate downstream signaling pathways through its effector MaRIC6, thereby coordinating root hair deformation and infection thread development during the early stages of rhizobial infection.
|
|
Scooped by
Jean-Michel Ané
December 25, 2025 7:20 PM
|
Many plant endosymbionts are facultative, switching between host-associated and free-living stages. Extensive genomic and experimental studies suggest that adaptation during the saprophytic, off-host phase, rather than adaptation to hosts, primarily constrains the biogeographic distribution of these microbes. To test this hypothesis, we analyzed the growth capacities and genomic features of 38 Sinorhizobium and Ensifer strains isolated from the nodules of Medicago lupulina (black medic), collected from two regions with distinct thermal environments. The warmer region is predominantly inhabited by S. meliloti, while S. medicae and Ensifer strains are more common in the cooler region. Laboratory assays demonstrated that at 40 °C, the upper temperature limit of their region of origin, S. meliloti remained viable, albeit with reduced growth, whereas S. medicae and Ensifer strains failed to grow under heat stress. Comparative genomics revealed isolation-by-distance in both the core and accessory genomes, particularly in S. meliloti in the warmer region, which exhibits less within-region thermal variation. This is consistent with an isolation-by-distance model where population divergence is governed by restricted gene flow. These findings suggest that metabolic constraints shape the regional distribution of this facultative microbial symbiont, while limited gene flow influences local population structure.
|