Your new post is loading...
Your new post is loading...

|
Scooped by
Gilbert C FAURE
July 11, 2024 4:21 AM
|
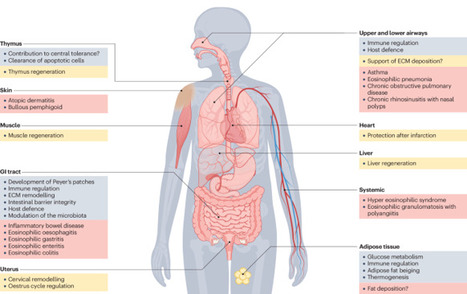
Eosinophils are bone marrow-derived granulocytes that are traditionally associated with type 2 immune responses, such as those that occur during parasite infections and allergy. Emerging evidence demonstrates the remarkable functional plasticity of this elusive cell type and its pleiotropic functions in diverse settings. Eosinophils broadly contribute to tissue homeostasis, host defence and immune regulation, predominantly at mucosal sites. The scope of their activities primarily reflects the breadth of their portfolio of secreted mediators, which range from cytotoxic cationic proteins and reactive oxygen species to multiple cytokines, chemokines and lipid mediators. Here, we comprehensively review basic eosinophil biology that is directly related to their activities in homeostasis, protective immunity, regeneration and cancer. We examine how dysregulation of these functions contributes to the physiopathology of a broad range of inflammatory diseases. Furthermore, we discuss recent findings regarding the tissue compartmentalization and adaptation of eosinophils, shedding light on the factors that likely drive their functional diversification within tissues. This Review by Arnold and Munitz discusses the diverse roles of eosinophils in the settings of tissue homeostasis, infection, allergy and cancer. The authors explain the molecular mechanisms that enable eosinophils to adapt to diverse tissue types and conditions, and they consider the therapeutic potential of eosinophil-depleting drugs in the clinic.
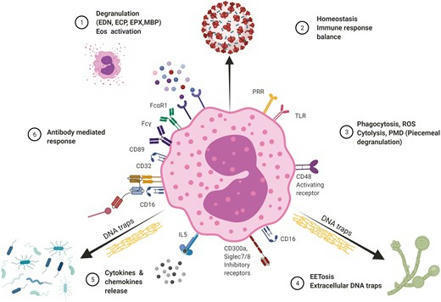
This review summarizes some of the updated information/recent findings on the role of eosinophil direct and antibody mediated interactions with pathogens.
|
|
Scooped by
Gilbert C FAURE
January 17, 2021 3:46 AM
|
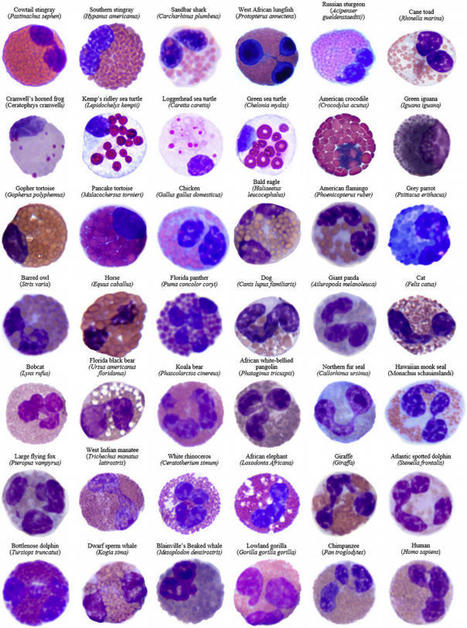
Eosinophils have distinctive morphological and biochemical features differentiating
them from other granulocytes. Fig 1 shows eosinophils in vertebrates across taxa from
an evolutionary perspective, demonstrating an appreciation of their morphological
diversity despite a number of conserved features that define the lineage. The presence
of “eosinophilic” or acidophilic blood cells/hemocytes has been recognized as well
in various invertebrate species, but it is still unknown whether these are evolutionary
precursors to eosinophils in vertebrates.
|
|
Scooped by
Gilbert C FAURE
September 25, 2020 4:07 AM
|
|
|
Scooped by
Gilbert C FAURE
December 5, 2019 9:04 AM
|
Eosinophils and their secretory mediators play an important role in the pathogenesis of infectious and inflammatory disorders.Although eosinophils are largely evolutionally conserved, their physiolog...
|
|
Scooped by
Gilbert C FAURE
November 17, 2018 5:24 AM
|
J Vet Med Sci. 2018 Nov 5. doi: 10.1292/jvms.18-0601.[Epub ahead of print]...
|
|
Scooped by
Gilbert C FAURE
July 13, 2018 4:44 AM
|
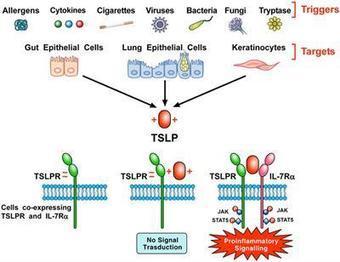
Thymic Stromal Lymphopoietin (TSLP) is a pleiotropic cytokine originally isolated from a murine thymic stromal cell line. TSLP exerts its biological effects by binding to a high affinity heteromeric complex composed of TSLPR chain and IL-7R. TSLP is primarily expressed by activated lung and intestinal epithelial cells, keratinocytes, and fibroblasts. However, dendritic cells, mast cells and presumably other immune cells can also produce TSLP. Different groups of investigators have demonstrated the existence of two variants for TSLP in human tissues: the main isoform expressed in steady state is the short form (sf TSLP), which plays a homeostatic role, whereas the long form (lfTSLP) is upregulated in inflammatory conditions. In addition, there is evidence that in pathological conditions TSLP can be cleaved by several endogenous proteases. Several cellular targets for TSLP have been identified, including immune (dendritic cells, ILC2, T and B cells, NKT and Treg cells, eosinophils, neutrophils, basophils, monocytes, mast cells, and macrophages) and non-immune cells (platelets and sensory neurons). TSLP has been originally implicated in a variety of allergic diseases (e.g., atopic dermatitis, bronchial asthma, eosinophilic esophagitis). Emerging evidence indicates that TSLP is also involved in chronic inflammatory (i.e., COPD, celiac disease) and autoimmune (e.g., psoriasis, rheumatoid arthritis) disorders and several cancers. These emerging observations greatly widen th
|
|
Scooped by
Gilbert C FAURE
March 23, 2017 2:54 AM
|
A human heart recuperating from inflammation, a condition called myocarditis, can take one of two paths: a healthy return to normal function or a dangerous
|
|
Scooped by
Gilbert C FAURE
October 19, 2015 1:47 AM
|
RT @britsocimm: "Eosinophils: important players in humoral immunity". Read full review: http://t.co/mpFeJUtDxq #immunology #science http://…
|
|
Scooped by
Gilbert C FAURE
September 14, 2014 2:39 PM
|
Peritumoral eosinophils predict recurrence in colorectal cancer Nature.com Mainly, the occurrence of lymphocytes was considered when assessing this lamina.21 In contrast, the grading scheme according to Klintrup et al,6 which was applied in the...
|
|
|
Scooped by
Gilbert C FAURE
September 25, 2023 10:26 AM
|
|
|
Scooped by
Gilbert C FAURE
August 2, 2021 4:00 AM
|
Th2 Cell Th2 cells stimulate B cell and eosinophil proliferation and reduce IFN-γ production by Th1 cells, thereby promoting humoral and allergic responses. From: Neurobiology of Disease, 2007 Related terms: View all Topics Effector CD4+ T Cells in the Intestines Craig L. Maynard, Casey T. Weaver, in Mucosal Immunology (Fourth Edition), 2015 Th2 Cells Th2 cells augment the eradication of parasitic helminthes that induce expression of IL-4 by innate immune cells, such as basophils and tissue-resident mast cells. IL-4 signaling to antigen-activated, previously naïve CD4 T cells results in activation of STAT6 and subsequent induction of the transcription factor GATA-3 (Bonecchi et al., 1998). Via secretion of IL-4, IL-5, and IL-13, Th2 cells orchestrate B cell class switching to IgE (Bonecchi et al., 1998), thereby priming basophils and mast cells for granule release, recruit eosinophils, and enhance mucus production, respectively. Human Th2 cells can be distinguished by surface expression of CCR4 and CRTH2 (Bonecchi et al., 1998; Abe et al., 1999; Nagata et al., 1999). Host Defenses in Skin Hui Xu, ... Craig A. Elmets, in Clinical Immunology (Fifth Edition), 2019 Th2 responses. Th2 cells are involved in type 2 immune responses, which are important for eradication of extracellular parasites and bacterial infection. They produce IL-4, IL-5, IL-10, and IL-13, which are important for the induction and development of humoral immune responses. IL-4 and IL-13 activate B-cell proliferation, Ig class-switching, and antibody production. Th2 cell-mediated inflammation is characterized by the presence of eosinophils and basophils, as well as extensive mast cell degranulation—a process dependent on cross-linking surface-bound IgE.24 IL-5 is a potent hematopoietic cytokine, which stimulates bone marrow production of eosinophils as well as activation and chemotaxis of eosinophils and basophils to affected tissue. In mice, Th2-cell deficiency profoundly increases susceptibility to Leishmania infection in skin. In humans, Th2 cells appear to play a critical role in the pathogenesis of atopic dermatitis (Chapter 44). A recent clinical trial with dupilumab, a fully human mAb that targets the IL-4 receptor-αα and blocks IL-4 and IL-13 signaling, improved atopic symptoms . Role of CD4+ T Cells in the Pathophysiology of Multiple Sclerosis Fumitaka Sato, ... Ikuo Tsunoda, in Multiple Sclerosis, 2016 Role of Th2 cells Th2 cells may play a protective role in MS, as Th2 immune responses have been shown to increase during remission in RRMS (Araki et al., 2003; Clerici et al., 2001). Decreased disease progression and exacerbation of MS during pregnancy have been associated with Th2-biased immune responses (Al-Shammri et al., 2004), although the exact mechanism remains unclear. Suppression of MS disease activities by immunomodulatory drugs, such as glatiramer acetate, has also been associated with enhanced Th2 immune responses (Weber et al., 2007). Experimentally, Th2 cells have been shown to regulate EAE and TMEV-IDD. In EAE induced with mouse spinal cord homogenate, injection of anti-IL-4 neutralizing mAb during the induction phase rendered resistant BALB/c mice susceptible to EAE (Constantinescu et al., 2001). The adoptive transfer of PLP-specific Th2 cell clones at the time of sensitization or disease onset prevented EAE in mice sensitized with PLP (Kuchroo et al., 1995). While T cell immunoglobulin mucindomain containing (TIM)2 has been shown to be preferentially expressed on the surface of Th2 cells and to negatively regulate Th2 immune responses, blockade of TIM-2/TIM-2 ligand interaction by administration of soluble TIM-2 fusion protein delayed the onset and decreased the severity of PLP-induced EAE by enhancing Th2 immune responses (Chakravarti et al., 2005). In TMEV-IDD, Th2 immune responses have also been demonstrated to suppress inflammatory demyelination in the CNS. Hill et al. (1998) demonstrated that during the early chronic phase of TMEV infection, infected mice treated with IL-4 developed less severe inflammatory demyelination compared with controls. Thus, the findings in EAE and TMEV-IDD suggest that Th1 cells could contribute to the pathogenesis of MS, while Th2 cells may play a protective role (Table 3). Cell-Mediated Defense against Infection Tobias M. Hohl, in Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases (Eighth Edition), 2015 Th2 Cells Th2 cells express a range of cytokines that influence B-cell differentiation and antibody production, eosinophil recruitment, and mucus production. The signature cytokines produced by Th2 cells are IL-4, IL-5, and IL-13, but Th2 cells can also produce IL-9, IL-10, IL-25, and amphiregulin.20 Th2 responses are generated when naïve T cells are exposed to IL-4 at the time of T-cell priming. In the setting of low antigen concentrations, IL-4 can be produced by responding T cells.21 After antigenic challenge, IL-4 can also be produced by mast cells and basophils in the vicinity of T-cell priming.22,23 IL-4 signals naïve T cells via the STAT6 pathway to express GATA3, the master regulator of Th2 differentiation,24 a process that can be enhanced by IL-4– and STAT6-independent GATA3 activation,25 all of which drives the expression of additional downstream activators. Although Th2 cells are best known for causing or contributing to allergic diseases such as atopic dermatitis, allergic rhinitis, and asthma, Th2 cells also contribute to defense against infections, particularly helminth infections of the gastrointestinal tract.26 In this setting, eosinophil recruitment, IgE production, and mucus hypersecretion can enhance parasite expulsion in an IL-4 and IL-13 signaling–dependent manner, a notion that is supported by murine studies of Nippostrongylus brasiliensis infection.27,28 The secretion of amphiregulin by Th2 cells can stimulate intestinal epithelial cell proliferation and expulsion of Trichuris muris, a nematode that infects mice.29 Besides Th2 cells, tissue-resident and Th2 cytokine-secreting innate lymphoid cells represent a significant source of IL-13 during the early stages of parasitic infection and promote expulsion.30-32 Aberrant Th2 responses to pathogens that require IFN-γ and Th1 responses for control can result in progressive infections and lethality. For example, Leishmania major infection of certain mouse strains induces Th2 responses that result in progressive in vivo replication and host death.33,34 In contrast, mouse strains that respond to L. major with Th1 responses clear and survive experimental infections. The mechanisms that determine whether an L. major–specific T-cell response will be predominately Th1 or Th2 are complex.35 In some mouse strains, Th2 responses occur because of T-cell responses to one dominant antigen called LACK (Leishmania analogue of the receptors of activated C kinase).36 In the absence of a T-cell response to this specific antigen, the responding CD4+ T cells differentiate into Th1 cells. In humans, the type of disease associated with Mycobacterium leprae infection is also tied to CD4+ T-cell differentiation. Th1 differentiation is associated with tuberculoid leprosy, a paucibacillary infection in which IFN-γ–producing T cells enhance microbial killing. The induction of type I interferon and IL-10 signaling in innate immune cells during leprosy can antagonize IFN-γ–dependent protection.37 Th2 differentiation is associated with high tissue densities of M. leprae and more robust, but ineffective, antibody responses.38,39 T Cells and Their Effector Functions Ruben C. Fragoso, ... Steven J. Burakoff, in Encyclopedia of Cancer (Second Edition), 2002 IV.B.2 Th2 T Cells Th2 cells promote IgE production and eosinophil function, which are the key players in the pathogenesis of allergic inflammation and immunity against parasitic infections. Cytokines such as IL-4 and IL-5 released by Th2 cells stimulate, respectively, B-cell switching to the production of IgE antibody and activation of eosinophils. The coordinate actions of these effector mechanisms result in heightened immunity against, for example, helminthic parasites, which can be coated with IgE and destroyed by the toxic granular contents of eosinophils. The balance between Th1 and Th2 cells may serve to determine the outcome of an infection. The Th1-mediated response is an effective deterrent for the protozoan parasite Leishmania major. In strains of mice with a genetic predisposition to mount predominately Th2 responses, infection by L. major results in a severe cutaneous and systemic disease that cannot be eliminated effectively. In contrast, if mice were vaccinized with leishmania antigens coadministered with IL-12 to induce a Th1 response, the mice are protected from subsequent challenges with L. major. In an analogous manner, responses to Mycobacterium leprae in humans can have two sharply different outcomes depending on the polarization of Th cells. In lepromatous leprosy, a Th2-dominated response can result in diffuse and destructive lesions due to an ineffective response against M. leprae antigens. In contrast, patients who develop a strong Th1-mediated immunity have a less destructive disease called tuberculoid leprosy. T-Cell Immunity Shannon A. Carty, ... Gary A. Koretzky, in Hematology (Seventh Edition), 2018 Th2 Cells Th2 cells are critical for the immune response against extracellular parasites, such as helminths, through production of IL-4, IL-5, and IL-13. At initial sites of parasitic infection, epithelial cells of the target organs, including the skin, lungs, and intestines, and resident cells of the innate immune system sense parasite-derived products and produce Th2-inducing cytokines, including thymic stromal lymphopoietin (TSLP), IL-4, IL-25, and IL-33. These cytokines then act on innate immune cells, including basophils and DCs, as well as directly on naive CD4+ cells to promote Th2 differentiation. Recent work has provided insight into how cytokine signaling, particularly IL-4 signaling, promotes Th2 differentiation. Through interaction with its receptor, IL-4 activates STAT6. STAT6 plays a vital role in Th2 differentiation, as evidenced by the profound reduction in development of this lineage in Stat6-deficient mice. STAT6 activation leads to its nuclear translocation and subsequent induction of the transcription factor GATA3, which, like T-bet for Th1 cells, is considered the master regulator of Th2 differentiation. GATA3 regulates Th2 cytokine production by binding and activating the “Th2 locus,” which includes the genes encoding IL-4, IL-5, and IL-13. When GATA3 function is abrogated, Th2 differentiation is virtually absent both in vitro and in vivo. In mature differentiated Th2 cells, GATA3 deficiency results in loss of IL-5 and IL-13 production. GATA3 is both necessary and sufficient for Th2 differentiation because forced expression either by retroviral constructs or transgenic expression promotes Th2 differentiation and represses Th1 differentiation. Repression of Th1 development occurs at least partially through GATA3-dependent inhibition of STAT4, thus interfering with Ifng gene transcription. TCR signal strength also is involved in determining if a naive T cell will differentiate into a Th1 or Th2 cell. Studies in mice using altered peptide ligands that have decreased affinity for particular TCRs and experiments using limiting doses of antigen have demonstrated that diminished TCR stimulation promotes Th2 cell differentiation. Differences in costimulation also affect Th2 pathway differentiation. Mice deficient in CD28 or its ligand have a more pronounced defect in Th2 responses, suggesting that these molecules may play a greater role in promoting Th2 differentiation than Th1 differentiation. IL-4 produced by mature Th2 cells acts in a positive feedback loop to promote further Th2 cell differentiation in naive T cells as they encounter antigen. Th2-derived IL-4 also mediates IgE class switching in B cells. Soluble IgE binds to and crosslinks its high-affinity receptor FcεRI on basophils and mast cells, promoting production of histamine and serotonin as well as several cytokines, including IL-4, IL-13, and TNF-α. IL-5 produced from Th2 cells recruits eosinophils, whereas Th2-derived IL-13 promotes both the expulsion of helminths during parasitic infection and also the induction of airway hypersensitivity. Th2 responses are critical for immunity against extracellular parasites, but excessive Th2 responses are associated with the pathologic conditions of allergy and airway hypersensitivity. The increase in asthma in the developed world has been linked to an imbalance of Th subsets with skewing toward “Th2-ness” in the population. Additional work is necessary to more firmly establish a molecular immunologic link to the epidemiology of these diseases. Chronic Inflammation and Atherosclerosis Jan Nilsson, ... Andreas Edsfeldt, in Early Vascular Aging (EVA), 2015 Interleukin-10 Th2 cells, Tregs, B-cells, monocytes, and macrophages are all potential sources of IL-10. The anti-inflammatory effects of IL-10 are mediated by inhibition of T-cell proliferation, macrophage apoptosis, antigen presentation, collagenase expression, and inflammatory cytokine production. In mice, IL-10 deficiency is associated with increased inflammatory cell invasion, a greater plaque burden, and an increased inflammatory cytokine response [40]. Human studies on circulating IL-10 revealed that high plasma levels of IL-10 are associated with an improved outcome and a lower risk for recurrent events in patients with acute coronary syndromes [41,42]. Group 2 Innate Lymphoid Cells in the Regulation of Immune Responses Ben Roediger, Wolfgang Weninger, in Advances in Immunology, 2015 7.8 IL-4/IL-4Rα Like Th2 cells, ILC2 cells express a functional IL-4 receptor (Doherty et al., 2012; Motomura et al., 2014), at least in the lung, and have been shown to produce IL-13 and IL-9 in response to IL-4 in vitro (Motomura et al., 2014). IL-4 was also shown to augment IL-2-driven proliferation of ILC2 cells in vitro (Motomura et al., 2014), which may relate to the STAT6 dependency of ILC2 cell proliferation in vivo (discussed further below). Animal Models of Immunity to Female Genital Tract Infections and Vaccine Development Charu Kaushic, ... Kenneth W. Beagley, in Mucosal Immunology (Fourth Edition), 2015 Th2 Cells CD4+ Th2 cells do not protect against chlamydial infection (Wang et al., 1999; Yang, 2001; Hawkins et al., 2002) and can exacerbate pathology (Chen et al., 2010; Wang et al., 1999; Perry et al., 1997) because of suppression of Th1 immunity. However, activation of Th2 cells is important for the production of IgG and IgA, both of which reduce infection in vivo. Th2 cells also may act as regulators of the Th1 response to limit tissue pathology after resolution of infection (Debattista et al., 2003). Indeed, it has been suggested that a human vaccine to prevent ascending infection and tissue inflammation should aim to elicit primarily a Th2 response to limit collateral damage (Vicetti Miguel and Cherpes, 2012). This approach would certainly be contrary to the current dogma driving vaccine research (see below).
|
|
Scooped by
Gilbert C FAURE
October 8, 2020 4:10 AM
|
Interleukin 10 Interleukin-10 (IL-10) is an important anti-inflammatory cytokine (Fiorentino, Zlotnik, Mosmann, Howard, & O’Garra, 1991; Fiorentino, Zlotnik, Vieira, et al., 1991). From: Advances in Cancer Research, 2015 Related terms: View all Topics Role of IL-10 and the IL-10 Receptor in Immune Responses A. Howes, ... A. O'Garra, in Reference Module in Biomedical Sciences, 2014 Conclusions and Unanswered Questions on IL-10 IL-10 is an anti-inflammatory cytokine that maintains the balance of the immune response, allowing the clearance of infection while minimizing damage to the host. IL-10 can also dampen the harmful immune responses elicited in autoimmunity and allergy. The consequence of this activity, however, is that IL-10 can contribute to chronic infection. The importance of IL-10 in this balance is supported by a wealth of evidence gathered from studies in both the human and mouse systems. The immune-stimulatory roles of IL-10 are less well understood but may be a factor in influencing the role of IL-10 in antitumor and/or mucosal immune responses while maintaining the response that limits immunopathology. Further understanding of the sources of IL-10 in various contexts, how IL-10 production is regulated in different cell types, and precisely which cells IL-10 in different immunological settings will greatly enhance our ability to use IL-10 as a potential immune therapy in the future. Introduction to Mechanisms of Allergic Diseases Terufumi Kubo, ... Cezmi A. Akdis, in Middleton's Allergy Essentials, 2017 Interleukin-10 (IL-10) IL-10 plays a role in the control of allergy and asthma. IL-10 inhibits many effector cells and disease processes, and its levels are inversely correlated with disease incidence and severity. IL-10 is synthesized by a wide range of cell types, including B cells, monocytes, DCs, NK cells, and T cells. It inhibits proinflammatory cytokine production and Th1 and Th2 cell activation, which is likely attributable to the effects of IL-10 on APCs and its direct effects on T cell function (Table 1-3). IL-10 levels inversely correlate with the incidence and severity of asthmatic disease in the lung. In addition, the levels of IL-10 inversely correlate with skin-prick test reactivity to allergens. Beekeepers, who undergo multiple bee stings and are naturally tolerant to bee venom allergen have a high IL-10 response. IL-10 and IL-10-producing Treg and Breg cells play essential roles in immune tolerance to allergens. In addition, the roles of Treg and Breg cells and IL-10 have been shown in many autoimmune, organ transplantation, tumor tolerance conditions.6 Interleukin-10 YaoZhong Ding, ... Jonathan Bromberg, in Encyclopedia of Hormones, 2003 V.A Inflammation IL-10 has protective effects in experimental endotoxemia and rescues mice from LPS-induced toxic shock, which is correlated with reduced levels of serum TNFα. IL-10 inhibits the production of TNFα and macrophage inflammatory protein-2 (MIP-2); regulates hemodynamic parameters, leukocyte–endothelial cell interactions, and microvascular permeability; and reduces mortality in experimental endotoxemia. Mice treated with anti-IL-10 from birth or IL-10-deficient mice are more susceptible to endotoxin-induced shock than are normal mice. Human volunteers receiving IL-10 after endotoxin challenge suffer fewer systemic symptoms and less cytokine production. Cytokines in GVHD and GVL Kate A. Markey, ... Geoffrey R. Hill, in Immune Biology of Allogeneic Hematopoietic Stem Cell Transplantation (Second Edition), 2019 Clinical Evidence for Interleukin-10 IL-10 polymorphisms were among the first to be correlated with disease outcome in GVHD [187]. The seminal paper by Lin and colleagues demonstrated a clear and dramatic association of recipient IL-10 genotype with GVHD outcome. Donor IL-10 genotype has also been associated with significantly lower risk of grades III–IV aGVHD [188]. The phenotypic correlation of these polymorphisms does remain uncertain however, i.e., it is not clear whether they lead to gain or loss of IL-10 function. It has been hypothesized that the protection from aGVHD is due to enhanced IL-10 production from APC [184]. INTERLEUKINS | IL-10 T.J. Standiford, J.C. Deng, in Encyclopedia of Respiratory Medicine, 2006 Regulation of Production and Activity IL-10 is produced by a variety of cell types, including CD8+ and CD4+ T cells (e.g., T-helper-2 (Th2) cells, regulatory T cells), γδ-T cells, NK cells, NK T cells, B cells, dendritic cells, eosinophils, mast cells, and monocytic cells (e.g., microglia, monocytes, macrophages). Macrophages are the major source of IL-10, although regulatory T cells are now recognized as an important subpopulation of T cells that release IL-10. Nonleukocyte cell populations, including epithelial cells, keratinocytes, and melanoma cells, also express IL-10. In the lung, human alveolar macrophages (AMs), bronchial epithelial cells, and alveolar epithelial cells have been shown to express IL-10 mRNA and protein. In contrast, murine resident AMs secrete minimal amounts of IL-10, although these cells do express IL-10 receptors. Macrophages produce IL-10 in response to inflammatory or infectious stimuli, including lipopolysaccharide, TNF-α, and catecholamines. High levels of IL-10 are also produced under conditions that induce anergy and tolerance, such as repeated antigen stimulation. In these contexts, IL-10 likely provides negative feedback to dampen the magnitude of immune responses. Interestingly, several viruses, such as EBV, produce IL-10 or IL-10-like molecules (vIL-10). The genomes of other viruses, including members of the herpesvirus family, poxvirus, and primate cytomegaloviruses (CMV), also contain homologs of the IL-10 gene. Many vIL-10 proteins share extensive sequence homology with human IL-10. Viral IL-10 appears to engage the same IL-10 receptor in the host and is capable of producing some of the same biological effects as host-derived IL-10. Given the anti-inflammatory properties of IL-10, vIL-10 likely confers a survival advantage for these viruses by suppressing antiviral host immune responses. Interleukin 10 Receptor Signaling Dror S. Shouval, ... Scott B. Snapper, in Advances in Immunology, 2014 Abstract Interleukin 10 (IL10) is a key anti-inflammatory cytokine that can inhibit proinflammatory responses of both innate and adaptive immune cells. An association between IL10 and intestinal mucosal homeostasis became clear with the discovery that IL10 and IL10 receptor (IL10R)-deficient mice develop spontaneous intestinal inflammation. Similarly, patients with deleterious mutations in IL10, IL10RA, or IL10RB present with severe enterocolitis within the first months of life. Here, we review recent findings on how IL10- and IL10R-dependent signaling modulates innate and adaptive immune responses in the murine gastrointestinal tract, with implications of their role in the prevention of inflammatory bowel disease (IBD). In addition, we discuss the impact of IL10 and IL10R signaling defects in humans and their relationship to very early-onset IBD (VEO-IBD). Spinal Cord Injury Samuel David, ... V. Wee Yong, in Handbook of Clinical Neurology, 2012 Interleukin 10 Interleukin 10 (IL-10) is a potent anti-inflammatory cytokine which reduces inflammation in several disease models. Several reports indicate that IL-10 promotes tissue protection and functional recovery after SCI. Mice lacking IL-10 showed worse functional outcome and greater inflammatory response, apoptosis, tissue loss, and edema after spinal cord compression (Genovese et al., 2009) or excitotoxic injury (Abraham et al., 2004). Furthermore, the increased tissue damage observed in IL-10 null mice, could be reversed with IL-10 administration (Abraham et al., 2004). In addition, a single injection of systemically administered IL-10 improved locomotor recovery and reduced TNF-α expression after spinal cord contusion injury in rats (Bethea et al., 1999). However, in another study, the same group failed to observe any protective effects of IL-10 after a similar type of lesion in different strain of rat (Takami et al., 2002). More animal studies are needed to test various doses, mode of delivery and timing of treatment to establish the role of IL-10 in SCI. Interleukin 10 and its Receptor Vijay P. Khatri, Michael A. Caligiuri, in Encyclopedia of Immunology (Second Edition), 1998 Cellular source of IL-10 IL-10 is produced by several types of cells, including activated T lymphocytes, monocytes, B cells as well as nonhematopoietic sources such as keratinocytes, colon carcinoma and melanoma cells. In humans though TH2 T cell clones are the main source of IL-10, many TH1 clones will also secrete IL-10 following antigen-specific stimulation. In contrast, mIL-10 is produced by the TH2 subset of CD4+ T cells but not by TH1 or CD8+ T cells. In humans both CD4+ and CD8+ T cells secrete IL-10 following stimulation with anti-CD3, although significantly higher levels are produced by CD4+ T cells. Among the CD4+ T cells, CD45RO+ memory cells produce 10-fold higher level of IL-10 than naive T-cells (CD45RA+). Murine Ly-1 B cells as well as Epstein–Barr virus transformed human B cells produce IL-10. Human monocytes are also a major source of IL-10 in response to activation with lipopolysaccharide. Interestingly, kinetics studies reveal that IL-10 is synthesized later than other immunoregulatory cytokines by activated T cells or monocytes. This suggests that IL-10 may play a regulatory role for the later phases of the immune response. The Immune Basis of Allergic Lung Disease Stefanie C.M. Burleson, ... Michael R. Van Scott, in Comparative Biology of the Normal Lung (Second Edition), 2015 IL-10 IL-10, originally described as “cytokine synthesis inhibitor factor,” is produced by TH0, TH1, TH2, Treg, cytotoxic T cells, mast cells, and activated monocytes. IL-10 receptors are located on lymphoid, myeloid, and NK cells. IL-10 decreases TH2 signaling by downregulating MHC class II, B7.1/B7.2 and CD23 expression (Bjorgo et al., 2011; Bopp et al., 2009; Baumer et al., 2007; Heijink and Van Oosterhout, 2006). IL-10 suppresses monocyte and macrophage activity, including release of ROI, and downregulates secretion of pro-inflammatory cytokines such as IL-1β, TNF-α, IL-6, IL-8, and MIP-1α. In TH1 cells, IFN-γ and IL-2 synthesis are inhibited, as is synthesis of IL-4 and IL-5 by TH2 cells. In contrast, IL-10 can promote growth of mast cells, T cells, and B cells. However, even these responses can be bimodal, with inhibition observed at high concentrations of IL-10. IL-10 mRNA and protein are reduced in alveolar macrophages from patients with asthma, although the number of IL-10 expressing T cells and macrophages may be increased. In Brown Norway rats, IL-10 is reported to attenuate late-phase responses and eosinophilia postchallenge. In mice, treatment with IL-10 at the time of sensitization decreases eosinophilia (Chung and Barnes, 1999). Studies involving antibodies against IL-10 and IL-10R, as well as IL-10 knockout, have revealed attenuation of AHR and eosinophilia. This apparent inconsistency in the typical TH1 functioning of IL-10 might be explained by the finding that IL-10 can inhibit IL-13Rα2, a protein responsible for binding and inhibiting IL-13, thereby potentially increasing IL-13 effects. It has been noted that mice lacking both IL-10 and IL-13Rα2 exhibit a more severe manifestation of pulmonary allergic disease than mice deficient in IL-10 or IL-13Rα2 alone (Barnes, 2008). Sepsis Hector R. Wong, ... Cláudio Flauzino de Oliveira, in Pediatric Critical Care (Fourth Edition), 2011 Interleukin-10 Interleukin-10 is the best studied and most well known antiinflammatory cytokine.40,41 As an antiinflammatory cytokine, IL-10 serves to antagonize the proinflammatory effects of other cytokines and can thereby keep inflammation “in check.” IL-10 inhibits expression of cytokines such as TNF-α, IL-1β, and IL-8, and can inhibit expression of adhesion molecules. In addition, IL-10 can “deactivate” monocytes by downregulating the expression of MHC surface molecules. Thus IL-10 has a number of interesting properties that could potentially be leveraged therapeutically to limit excessive inflammation during sepsis. This theoretical consideration must be tempered by the ability of IL-10 to deactivate monocytes and thereby potentially impair the ability to adequately clear infection (i.e., the immune suppression paradigm depicted in Figure 103-1). Indeed, it has been reported that in children with multiple organ dysfunction syndrome, higher plasma IL-10 levels correlate with higher mortality, and higher monocyte mRNA levels of IL-10 correlate with increased length of stay in the ICU.42 Similar observations have been reported in adult patients with septic shock.43
|
|
Scooped by
Gilbert C FAURE
July 27, 2020 8:22 AM
|
Recent studies have provided insights into the pathogenesis of coronavirus disease 2019 (COVID-19)1–4. Yet, longitudinal immunological correlates of disease outcome remain unclear. Here, we serially analysed immune responses in 113 COVID-19 patients with moderate (non-ICU) and severe (ICU) disease. Immune profiling revealed an overall increase in innate cell lineages with a concomitant reduction in T cell number. We identify an association between early, elevated cytokines and worse disease outcomes. Following an early increase in cytokines, COVID-19 patients with moderate disease displayed a progressive reduction in type-1 (antiviral) and type-3 (antifungal) responses. In contrast, patients with severe disease maintained these elevated responses throughout the course of disease. Moreover, severe disease was accompanied by an increase in multiple type 2 (anti-helminths) effectors including, IL-5, IL-13, IgE and eosinophils. Unsupervised clustering analysis identified 4 immune signatures, representing (A) growth factors, (B) type-2/3 cytokines, (C) mixed type-1/2/3 cytokines, and (D) chemokines that correlated with three distinct disease trajectories of patients. The immune profile of patients who recovered with moderate disease was enriched in tissue reparative growth factor signature (A), while the profile for those with worsened disease trajectory had elevated levels of all four signatures. Thus, we identified development of a maladapted immune response profile associated with severe COVID-19 outcome and early immune signatures that correlate with divergent disease trajectories.

|
Suggested by
Société Francaise d'Immunologie
July 9, 2019 1:33 PM
|
Abstract Eosinophilic leukocytes develop in the bone marrow and migrate from peripheral blood to tissues, where they maintain homeostasis and promote dysfunction via release of preformed immunomodulatory mediators. In this study, we explore human eosinophil heterogeneity with a specific focus on naturally occurring variations in cytokine content. We found that human eosinophil-associated cytokines varied on a continuum from minimally (coefficient of variation [CV] ≤ 50%) to moderately variable (50% < CV ≤ 90%). Within the moderately variable group, we detected immunoreactive IL-27 (953 ± 504 pg/mg lysate), a mediator not previously associated with human eosinophils. However, our major finding was the distinct and profound variability of eosinophil-associated IL-16 (CV = 103%). Interestingly, eosinophil IL-16 content correlated directly with body mass index (R2 = 0.60, ***p < 0.0001) in one donor subset. We found no direct correlation between eosinophil IL-16 content and donor age, sex, total leukocytes, lymphocytes, or eosinophils (cells per microliter), nor was there any relationship between IL-16 content and the characterized −295T/C IL-16 promoter polymorphism. Likewise, although eosinophil IL-1β, IL-1α, and IL-6 levels correlated with one another, there was no direct association between any of these cytokines and eosinophil IL-16 content. Finally, a moderate increase in total dietary fat resulted in a 2.7-fold reduction in eosinophil IL-16 content among C57BL/6-IL5tg mice. Overall, these results suggest that relationships between energy metabolism, eosinophils, and IL-16 content are not direct or straightforward. Nonetheless, given our current understanding of the connections between asthma and obesity, these findings suggest important eosinophil-focused directions for further exploration.
|
|
Scooped by
Gilbert C FAURE
September 17, 2018 4:20 AM
|
Review Free access | 10.1172/JCI90962 The role of the complement system in cancer Vahid Afshar-Kharghan First published March 1, 2017 - More info Abstract In addition to being a component of innate immunity and an ancient defense mechanism against invading pathogens, complement activation also participates in the adaptive immune response, inflammation, hemostasis, embryogenesis, and organ repair and development. Activation of the complement system via classical, lectin, or alternative pathways generates anaphylatoxins (C3a and C5a) and membrane attack complex (C5b-9) and opsonizes targeted cells. Complement activation end products and their receptors mediate cell-cell interactions that regulate several biological functions in the extravascular tissue. Signaling of anaphylatoxin receptors or assembly of membrane attack complex promotes cell dedifferentiation, proliferation, and migration in addition to reducing apoptosis. As a result, complement activation in the tumor microenvironment enhances tumor growth and increases metastasis. In this Review, I discuss immune and nonimmune functions of complement proteins and the tumor-promoting effect of complement activation. Introduction The complement system is a cascade of serine proteases encoded by genes originating from the same ancestral genes as coagulation proteins (1). Like the coagulation system, complement activation involves several steps, is tightly regulated, and requires both plasma and membrane proteins (2, 3). Many complement proteins possess dual functions that provide crosstalk between the complement system and other effector and regulatory systems. As a result, the complement system participates in adaptive immunity, hemostasis, neuroprotection and synaptic pruning, and organ development in addition to its role in innate immunity. It is also involved in a diverse array of pathologic conditions, such as thrombotic disorders, autoimmune disorders, schizophrenia, alloimmune responses including allograft rejection and graft-versus-host disease, and cancer. The complement system’s role in fighting invasive pathogens has been extensively studied (4, 5), but recent discoveries provide new perspectives on the complement system’s function in the extravascular and interstitial tissue compartment. These discoveries illustrate an important role for complement proteins in cell-cell and stroma-cell communications. In this Review, I briefly discuss activation, regulation, immune, and nonimmune functions of the complement system to provide a framework for examining the role of complement in cancer. Activation of the complement system The complement system is activated by three major pathways: the classical pathway, via antigen-antibody complexes; the alternative pathway, via any permissive surfaces; and the lectin pathway, via binding of pattern-recognizing mannose-binding lectins (MBLs) to carbohydrate ligands on the surface of pathogens (Figure 1 and refs. 6–9). The convergence point for all complement activation pathways is the formation of the C3 convertase complex on the surface of targeted cells, summarized in Figure 1, A–C. After forming C3 convertase, complement is able to carry out its effector functions. Figure 1 Complement activation. (A) The classical pathway is initiated by a complement-fixing antibody binding to an antigen on targeted cells. C1q binds to the antibody’s Fc domain in the antibody-antigen complex. C1r and C1s assemble on C1q, C1r cleaves and activates C1s, and activated C1s cleaves C4 and C2 into C4b and C2a, respectively. C4b and C2a form the C3 convertase C4bC2a. (B) In the lectin pathway, MBL binds to repetitive sugar moieties such as mannose. MBL and MASP2 then form a C1-like complex. Activated MASP2 in MBL-MASP2 complex cleaves C4 and C2 and generates C3 convertase (C4bC2a). (C) In the alternative pathway, small amounts of hydrolyzed plasma C3 [C3(H2O)] bind to factor B, which forms the C3(H2O)Bb complex with help from factor D. C3(H2O)Bb cleaves additional plasma C3 to generate highly active C3b, which binds to cell the surface. On a complement-activating surface, C3b binds Bb (produced by factor D–mediated cleavage of factor B) and generates C3bBb (the alternative pathway’s C3 convertase). (D) Regardless of the initiation steps, C3 convertase deposits additional C3b molecules and generates C3a. If it remains intact, C3 convertase binds to additional C3b to generate C5 convertase. C5 convertase cleaves C5 to generate C5b. (E) C5b binds to C6, C7, and C8, forming a C5b-8 complex, which polymerizes several C9 molecules, forming the cytolytic MAC. In all three complement activation pathways, C3 convertase complex cleaves C3 molecules to C3a, one of the two major anaphylatoxins, and to C3b, a potent opsonin. Binding of C3b molecules to the surface of cells or cell debris in a process called opsonization marks them for phagocytosis by macrophages. Surface-bound C3b and its degradation products are ligands for complement receptors CR1, CR3, and CR2 that are expressed on myelomonocytic cells, lymphocytes, and follicular dendritic cells. Binding of C3b and its degradation products to correspondent receptors are crucial to cell-cell interactions in the innate and adaptive immune responses and in the removal of complement-coated apoptotic and necrotic cells. Propagation of complement activation by C3 convertase results in the generation of the C5 convertase complex on the cell surface. C5 convertase then cleaves C5 to C5a and C5b. C5a is a potent anaphylatoxin and recruits neutrophils to areas of inflammation and tissue damage. C5b forms a complex with C6 and C7 that may insert into cell membrane, and subsequently be joined by C8 and multiple C9 to form the membrane attack complex (MAC or C5b-9 complex; Figure 1D). Deposition of an adequate number of MACs disrupts the phospholipid bilayer of the cell membrane, leading to massive calcium influx, loss of mitochondrial membrane potential, and cell lysis. However, MAC deposition at sublytic concentrations on cell membrane has a different result, activating intracellular signal transduction and cell proliferation (10). Eukaryotic cells have developed several defense mechanisms to counteract the dire consequences of MAC accumulation at the cell surface, including expression of complement regulatory proteins (CRPs) that disassemble MAC (i.e., CD59, vitronectin, and clusterin), and endocytosis or shedding of MAC from the cell surface. Thus, the three main consequences of complement activation are tagging of cells by C3b degradation products for phagocytosis; chemotaxis of inflammatory cells in response to C3a and C5a; and MAC-mediated cell lysis. As described below, complement activation end products affect tumor growth by altering cancer cell behavior and modulating the immune response to the tumor. Regulation of the complement system The complement system’s ability to cause cellular damage is strictly controlled at several steps, both in the fluid phase and on the cell surface (6). In the classical and lectin pathways, C1 inhibitor (C1INH) binds to and inactivates C1r, C1s, and MBL-associated serine proteases (MASPs). The activities of other CRPs can be categorized into two major groups: (a) decay-accelerating activity, which breaks up the C3 convertase complex, as can be seen in C4-binding protein (C4bp), CR1, decay-accelerating factor (DAF, also known as CD55), and factor H; and (b) membrane cofactor activity, which acts as a cofactor for the factor I–mediated cleavage of C3b or C4b to their inactive degradation products, iC3b and iC4b, respectively. CRPs with membrane cofactor activity include C4bp, CR1, membrane cofactor protein (MCP, or CD46), and factor H. Another important CRP is CD59, which is expressed on many different cell types and prevents assembly of MAC on the cell membrane. The anaphylatoxins C3a and C5a are complement activation products that are rapidly inactivated in plasma by carboxypeptidases, particularly carboxypeptidase N (11). CRPs are overexpressed by many cancer cells and may be used as potential therapeutic targets. Immune function of the complement system The complement system is an ancient defense mechanism preceding adaptive immunity (12). It can be activated by pattern-recognition molecules and natural antibodies (13). Complement system activation and generation of anaphylatoxins orchestrate an inflammatory response to pathogens (12, 14). Anaphylatoxins activate macrophages, neutrophils, mast cells, basophils, and eosinophils, resulting in their degranulation and the production of cytokines, which in turn causes vasodilation, increases vascular permeability, and enhances neutrophil extravasation and chemotaxis (13). The complement system links innate immunity to adaptive immunity. Complement deficiency impairs both B and T cell responses (15). The effect of complement on the B cell response is mediated by CR2 on B cells and follicular dendritic cells. Activation of the classical pathway on the surface of an antigen tags that antigen with C3d, enabling its binding to CR2 on B cells. CR2, CD19, and CD81 form a B cell coreceptor complex, and CR2 engagement with C3d enhances signaling through antigen-encountered B cell receptors and decreases the activation threshold of B cells (12, 15). The interaction between CR2 on follicular dendritic cells and C3d on antigens is important for antigen presentation to naive and primed B cells in the germinal center of lymph nodes, in the maturation of B cells, and in the generation of memory B cells. The role of complement proteins in the cognate interaction between antigen-presenting cells (APCs) and T cells is important in the T cell immune response (16). In addition to systemic production in the liver, complement proteins are also produced locally by T cells and APCs (17–20). The effects of complement proteins on activation, proliferation, and differentiation of T cells are mediated by the local complement activation, by production of C3a and C5a at the interface of T cells and APCs, and through anaphylatoxin receptors on T cells and APCs (17–19, 21, 22). Reducing the number of C3a and C5a receptors (C3aR and C5aR, respectively) on T cells or APCs impairs T cell immunity. Complement proteins and receptors are involved in different stages of the interaction between APCs and T cells. APCs produce C3 and express C3aR and C5aR, both of which are essential for their maturation and differentiation (19) and for effective antigen presentation to T cells (17, 23, 24). C3- or C3aR-deficient APCs are much less potent in inducing a T cell immune response compared with WT APCs (19, 25). After APCs present antigen to T cells, C5aR on the T cells is required for their proliferation. Binding of C5a to C5aR on T cells has both antiapoptotic and pro-proliferative effects (22). Nonimmune function of the complement system Cell-cell and stroma-cell interactions mediated by complement proteins regulate several physiologic processes, such as collective cell migration during embryogenesis (26), synaptic pruning during brain development (27–30), cell proliferation and differentiation during liver regeneration (31) and bone development (32, 33), and hematopoietic stem cell migration and engraftment during hematopoiesis (34). Complement and cancer The surge of interest in cancer immunotherapy is mainly focused on manipulating function or number of cytotoxic T cells. However, two important reasons justify studying the role of complement activation in cancer progression and the effect of complement manipulation in cancer therapy. First, the complement system is an important component of the inflammatory response, and inflammation is involved in various stages of tumorigenesis and cancer progression (35). Second, complement activation regulates adaptive immune response (15) and might have a role in regulating T cell response to tumors. Complement system in inflammation and tumorigenesis. Tumor-promoting inflammation has an important role in carcinogenesis and cancer progression (36–38). A series of elegant experiments established that activation of the complement system is an important component of tumor-promoting inflammation. Bonavita et al. showed that C3-deficient mice were protected against chemical carcinogenesis in mesenchymal and epithelial tissues (39), mainly because of reduced inflammation. Authors identified a humoral component of innate immunity, the long pentraxin PTX3, as an important negative regulator of inflammation and complement activation. PTX3-deficient mice were susceptible to chemical carcinogenesis, displaying an increased number of tumor-associated macrophages with M2 phenotype and increased concentration of CCL2 chemokine inside tumors. The tumor-promoting inflammation induced by PTX3 deficiency was complement-dependent and completely reversed after removal of C3, as manifested by a reduction in the susceptibility of Ptx3–/– C3–/– mice to chemical carcinogenesis. Similarly, treatment with the C5aR antagonist PMX-53 reversed the susceptible phenotype of Ptx3–/– mice without affecting the rate of tumorigenesis in Ptx3+/+ mice. Activation and regulation of complement pathways in tumors. Expression of complement and CRPs is increased in malignant tumors and cancer cell lines (summarized in Table 1). Complement proteins, C3 degradation products, and complement activation products (i.e., C5a, C3a, and C5b-9) are easily detectable in various types of cancer, consistent with complement activation inside these tumors. Table 1 Complement proteins in cancer The main pathway involved in activation of complement inside tumors is unclear, and evidence supports activation of each complement pathway in malignant tumors (40). To make matters more complicated, cancer cell membrane-bound serine proteases can also cleave C5 and generate C5a without complement activation (41). Additionally, complement proteins expressed in tumors might also play a role in cancer progression independent of complement activation, as was shown for C1q in a syngeneic murine model of melanoma, where C1q expression affected angiogenesis, tumor progression, and metastasis (42). In this murine model, C1q was expressed in endothelial cells, spindle-shaped fibroblasts, and tumor-infiltrating myeloid cells independently of C4. Lack of C4 coexpression in C1q-expressing tumors hints at a role for C1q in tumor progression independent of the classical pathway. Expression of CRPs, including both membrane proteins (CD55, CD59, MCP, or CD46) and soluble proteins (factor H and factor H–like proteins), is increased in cancer cells (43), although the overexpression of CRPs is heterogeneous among different cancer types and even between different tumor specimens of the same type of cancer (44). One interpretation of the presence of both complement activation products and CRPs in tumors is that complement activation is a host defense mechanism against cancer, and cancer cells resist complement attack by overexpressing CRPs. However, as discussed later in this Review, several recent studies do not support this interpretation and suggest another scenario in which local complement activation inside tumors enhances tumor growth. Complement activation: antitumor or protumor? Evidence for the effects of complement on malignant transformation of epithelial cells and progression of cancer has evolved based on several recent studies showing complex and sometimes contradictory findings. This complexity is similar to the complex role of inflammation in cancer (45). Although inflammatory cells and cytokines are important in immune surveillance, exemplified by the benefit of bacillus Calmette-Guérin therapy in early stages of bladder cancer, chronic inflammation promotes carcinogenesis and tumor growth. Even immune cells, such as macrophages, can have both pro- and antitumor phenotypes. Despite this multifaceted picture, most evidence points toward a protumor effect of chronic inflammation (45). The long-held view of complement activation as an antitumor defense mechanism is based on two main concepts: first, the complement system’s participation in immune surveillance against malignant cells, and second, complement-dependent cytotoxicity of therapeutic monoclonal antibodies. I will discuss these concepts below, and summarize new information pointing toward a protumor effect of complement activation inside tumors. Complement and immune surveillance. The complement system’s ability to distinguish self from non-self makes it an important part of the innate immune response to invading pathogens (46). Expression of non-self antigens and lack of CRPs on microbes make them optimal targets for complement detection and, later on, complement-mediated elimination. Similarly, expression of danger signals and neoantigens by apoptotic cells and cellular debris optimizes their detection and removal by the complement system. Cancer cells, on the other hand, mostly express the same proteins as their normal epithelial cell counterparts, albeit occasionally with a different density. Furthermore, overexpression of CRPs by cancer cells limits immune surveillance by the complement system (3, 43, 46, 47). Putting these findings together, one can conclude that cell-mediated immunity plays a more important role than humoral immunity in immune surveillance against cancer cells (48, 49), and effectiveness of complement in early detection and elimination of cancer cells is uncertain (50). Complement-dependent cytotoxicity. Complement activation was considered detrimental to cancer cells via complement-dependent cytotoxicity, which causes cancer cell lysis via MAC accumulation or phagocytosis of opsonized cancer cells by macrophages and neutrophils. Complement-dependent cytotoxicity is considered to be the main mechanism for the effectiveness of antitumor monoclonal antibodies. Rituximab, an anti-CD20 antibody against malignant B cells, is among the oldest and most widely used therapeutic monoclonal antibodies. Although in vitro and in vivo studies show that rituximab activates the classical complement pathway (51, 52), the notion that its therapeutic benefits are mainly mediated by induction of complement attack on malignant B cells is questionable. In fact, the antitumor effect of rituximab was inhibited by deposited complement proteins on B cells (53), and was enhanced in complement-deficient mice (54). Therefore, the extent to which complement-dependent cytotoxicity contributes to other immunologic effects of rituximab, i.e., antibody-dependent cellular cytotoxicity and antibody-dependent phagocytosis, is unknown. Other studies on the therapeutic mechanism of rituximab also showed a complement-independent, proapoptotic effect mediated by cross-linking of CD20 (55), as well as antiproliferative and antisurvival effects that were mediated by inhibition of B cell receptors (56). Furthermore, many in vitro antitumor effects of complement-fixing antibodies on cancer cell lines were not reproduced in vivo (57). Complement activation promotes tumor growth. Considering that complement is not efficient in immune surveillance against cancer cells and that the main antitumor effect of monoclonal antibodies might not arise from complement activation, the data supporting an antitumor role for complement activation are scant. The question remains: If complement does not attack cancer cells, how does local complement activation and deposition of complement proteins affect tumors? To understand the consequence of complement activation inside tumors, it is helpful to reexamine the biological functions of complement activation products. C3b and its degradation products binding to CR1, CR2, and CR3 provide ligands and receptors for cell-cell and stroma-cell interactions in many physiologic and pathologic conditions. Complement activation generates C3a and C5a and MAC. The anaphylatoxin receptors C3aR and C5aR are G protein–coupled receptors present on many cell types, including lymphocytes, monocytes/macrophages, myeloid cells, hematopoietic stem cells, mesenchymal cells, and epithelial cells, including cancer cells. Anaphylatoxin receptor signaling has been studied extensively (58). Activation of C5aR promotes a range of responses depending on the cell type. Relevant to its role in cancer, C5aR activation generates prosurvival and antiapoptotic responses. For example, C5a binding to C5aR decreases apoptosis in neutrophils (59) and T cells (22), and increases cell proliferation in endothelial (60) and colon cancer cell lines (61). Activation of C3aR plays an important role in guiding collective cell migration (26) and epithelial-mesenchymal transition (62, 63), both important mechanisms in metastasis. In a sublytic density, MAC accumulation on the cell membrane promotes cell proliferation (64) and differentiation, inhibits apoptosis (10, 65), and protects cells against complement-mediated lysis (66). Markiewski et al. showed that the activation of the classical complement pathway inside implanted orthotopic tumors in mice enhanced tumor growth (67). Complement’s progrowth effect on tumors was C5a-dependent and was eliminated in C5aR-deficient mice and in WT mice treated with a C5aR antagonist. C5a modulates the immune response to tumors by acting as a chemotactic factor, increasing infiltration of myeloid-derived suppressor cells (MDSCs) and reducing the number of CD8+ cytotoxic T cells inside tumors. MDSCs are immature myeloid cells that increase in blood, bone marrow, and spleen of tumor-bearing mice and cancer patients (68, 69) and assist tumor cells in evading the antitumor immune response. MDSCs reduce proliferation and increase apoptosis in CD8+ T cells by generating ROS and reactive nitrogen species (70). Depletion of CD8+ T cells in mice eliminated the protective effect of complement deficiency against tumor growth. In summary, this study showed that the immunomodulatory effect of activated classical complement pathway inside tumors enhances tumor growth. The origin of complement proteins was the host, but activation of complement occurred inside the tumor microenvironment, and the final effect on the tumor was an indirect immunomodulatory effect mediated by MDSCs (Figure 2). Figure 2 Effect of complement activation in the tumor microenvironment. Activation of the complement system inside tumors releases C5a and C3a into the tumor microenvironment and promotes tumor growth. C5a attracts myeloid cell, including MDSCs, into the tumor. MDSCs then reduce cytotoxic T cell responses to the tumor by inducing apoptosis and inhibiting CD8+ TILs via generation of ROS and reactive nitrogen species and depletion of arginine. In melanoma, secretion of C3 by CD8+ TILs and complement activation in the vicinity of these cells reduce IL-10 production by TILs and inhibit their function. Some cancer cell types secrete complement proteins into the tumor microenvironment and initiate an autocrine loop that increases cell proliferation and promotes metastasis. The effect of complement activation on MDSCs, TILs, and cancer cells is mediated by the C5a and C3a receptors (C5aR and C3aR) on these cells. In a follow-up study, Nunez-Cruz et al. investigated complement’s role in tumorigenesis in a murine model of spontaneous ovarian cancer (71, 72). C3 or C5aR deficiency in these mice prevented the development of ovarian tumors, permitting no tumors or only small and poorly vascularized tumor formation (71). C3 deficiency was associated with a change in the immune profile of leukocytes infiltrating into the tumors, but C5aR deficiency reduced ovarian tumor size without altering the immune profile of infiltrating leukocytes. This result suggested the existence of a protumor effect of complement that was independent of its immunomodulatory effect. We investigated the effect of complement in murine models of ovarian cancer and confirmed activation of complement in the tumor microenvironment (73). However, complement proteins detected inside ovarian tumors originated not from the host, but from tumor cells themselves. Complement activation products were present even inside tumors implanted in C3-deficient mice lacking a functional complement system. Although orthotopic ovarian cancer tumors in C3-deficient mice reached to the same size as those in WT mice, reducing C3 or C5 production in cancer cells significantly reduced the tumor growth independent of the host’s complement sufficiency status. C3 synthesis can be detected in malignant epithelial cells originating from several different organs, particularly lung and ovary. Inhibiting synthesis of complement proteins in cancer cells altered the immune profile of leukocytes infiltrating into tumors, manifested by an increase in the number of CD8+ T cells and reduction in myeloid cells. However, immunomodulatory effect of complement inhibition was not the main mechanism responsible for the observed reduction in tumor growth. Inhibiting complement protein synthesis in cancer cells implanted in CD8+ T cell–deficient mice reduced tumor growth to the same magnitude as in WT mice. We investigated the possibility of an autocrine stimulation of cancer cells as a result of complement activation. Anaphylatoxin receptors are present on ovarian cancer cells, and stimulation of these receptors by C3a or C5a agonist peptides increased proliferation and invasiveness of ovarian cancer cells in vitro. Furthermore, knockdown of these receptors on cancer cells reduced growth of orthotopic ovarian tumors in mice. Our studies showed that local complement activation inside the tumor microenvironment enhances tumor growth via a direct autocrine effect on ovarian cancer cells increasing cell proliferation (Figure 2). In a murine model, Wang et al. reported another mechanism for the progrowth effect of complement activation in melanoma, showing that production of IL-10 by CD8+ tumor-infiltrating lymphocytes (TILs) is constitutively inhibited in an autocrine fashion by C3 originating from CD8+ TILs themselves, acting through C5aR and C3aR on the surface of these lymphocytes (74). C3aR and C5aR antagonists increased IL-10 production and activated CD8+ TILs that in turn reduce tumor growth. The IL-10–dependent antitumor activity of complement inhibitors in melanoma was independent of the PD-1/PD-L1 axis or MDSCs. This study provides evidence that local complement activation in the tumor microenvironment results in suppression of the immune response to melanoma by inhibiting CD8+ TIL function (Figure 2). The studies above describe different mechanisms by which complement activation in the tumor microenvironment can enhance tumor growth: (a) by altering the immune profile of tumor-infiltrating leukocytes, (b) by increasing cancer cell proliferation, and (c) by directly suppressing CD8+ TIL function. It is possible that different cancer types use different mechanisms to take advantage of ectopic complement activation inside tumors. For example, ovarian cancer cells synthesize a significant amount of complement proteins and initiate an autocrine loop resulting in increased cell proliferation by a direct effect of anaphylatoxins on cancer cells. Conversely, melanoma cells do not secrete complement proteins, and complement proteins produced by CD8+ TILs reduce their IL-10 production and antitumor activity. An important question remains whether complement activation has any role in malignant transformation of normal cells or only affects the expansion of already established malignant clones. Most available data are based on orthotopic murine models of ovarian cancer or mice genetically engineered to develop ovarian cancer by overexpression of oncogenes. These studies showed that complement promotes growth and expansion of malignant tumors. Bonavita et al. showed that complement promotes malignant transformation of cells exposed to chronic inflammation induced by chemical carcinogens (39). However, additional studies are required to dissect the effect of early versus late stages of complement activation on various stages of oncogenesis. Overexpression of CRPs on cancer cells If complement activation promotes tumor growth and oncogenesis, why are CRPs overexpressed on cancer cells? One would expect that cancer cells, under selective pressure, downregulate expression of CRPs to benefit from complement activation. To reconcile these seemingly counterintuitive observations, we put forward the following hypothesis: Anaphylatoxins and sublytic concentrations of MAC promote tumor growth, but higher concentrations of MAC have a tumoricidal effect. As a result, cancer cells benefit from early stages of complement activation and production of anaphylatoxins, but actively inhibit generation of MAC. Cancer cells reduce MAC concentration by overexpressing CD59, the most consistently overexpressed CRP on different cancer cells (75) and the most effective membrane regulatory protein against complement-mediated lysis (43, 76, 77), eliminating MAC from the cell surface through membrane vesiculation. Thus, from a therapeutic point of view, interventions that reduce complement activation or promote the generation of MAC inside tumors can be considered as logical options to counter the progrowth effect of complement on cancer. Complement activation in epithelial-mesenchymal transition Epithelial-mesenchymal transition (EMT) occurs in physiologic processes such as embryogenesis and organ development, and in pathologic conditions including tissue fibrosis and metastasis (78, 79). Complement participates in EMT in murine models of renal injury and fibrosis (62, 80, 81). We showed that complement activation inside tumors not only increases tumor growth but also enhances metastasis by promoting EMT in cancer cells. In ovarian cancer cells, the transcription factor TWIST1 upregulates C3 gene expression, generating C3a in the tumor microenvironment, which binds to C3aR on ovarian cancer cells. We further showed that C3aR signaling increases EMT and decreases E-cadherin expression in ovarian cancer cells via a Krüppel-like factor 5–dependent mechanism and promotes EMT and metastasis (63). In addition to promoting metastasis, EMT also induces resistance to complement-dependent cytotoxicity in lung cancer cells by increasing expression of CD59 (82). Inhibition or knockdown of CD59 restored sensitivity of cancer cells to complement-dependent lysis without altering the morphologic features or protein markers of EMT in these cells. Therapeutic potential of targeting complement activation in cancer Our understanding of the role of the complement system in cancer biology is evolving, changing our approach to the therapeutic use of reagents modifying the complement system. Traditional methods targeting cancer cells using antitumor antibodies to promote lysis of cancer cells by MACs require identification of tumor-specific antigens that either are expressed with a higher density on cancer cells than normal epithelial cells, or are only expressed on cancer cells. Antibodies against EGFR and CD20 are among the most successful therapeutic antibodies. Development of therapeutic antibodies was initially complicated by induction of an immune response to polyclonal antibodies developed in nonhuman hosts. Development of monoclonal murine antibodies; later, chimeric human-mouse antibodies; and recently, humanized antibodies (83) helps overcome this problem. However, a more important problem in harvesting complement-dependent cytotoxicity induced by therapeutic antibodies is overexpression of membrane CRPs by cancer cells that let cancer cells evade MAC-mediated cytolysis (43). As a result, blockade of membrane CRPs on cancer cells, alone or in conjunction with use of therapeutic antibodies, has been tried as another potential therapeutic strategy. Blocking CRPs reduces cancer cell proliferation in vitro (43) and tumor growth in mice (76). CD59 blocking antibodies or CD59 siRNA enhanced complement-mediated cytolysis induced by anti-EGFR monoclonal antibodies (trastuzumab and cetuximab) in human lung cancer cell lines (84). rILYd4, a recombinant protein inhibitor of CD59, increased sensitivity of malignant B cells to rituximab in vitro and in orthotopic murine models (85). Membrane CRPs are universally expressed, and an important theoretical complication of blocking CRPs is exposing normal cells to complement-dependent cytotoxicity. For example, CD59 and CD55 protect red blood cells against complement-induced hemolysis, and blocking CD59 might cause hemolysis. Interestingly, administration of rILYd4 in mice was not associated with significant increases in hemolysis (85). More recent studies showed a protumor effect of complement, and inhibition of complement activation in vitro or in murine models of cancer was investigated as a novel way to treat cancer. Understanding of the autocrine and paracrine effects of complement production and activation inside tumors versus its systemic immunomodulatory effect is not complete, but blocking complement activation or inhibiting C5aR and C3aR signaling inside tumors seems a reasonable approach. However, several questions and concerns regarding the therapeutic use of anticomplement reagents have not been addressed and require additional studies: Does systemic complement inhibition affect local production and activation of complement in the tumor microenvironment? Pharmacokinetic studies based on measurements of tissue concentration of various anticomplement reagents may resolve this issue, although the leakiness of tumor vasculature likely provides adequate tissue penetrance of these reagents. Does complement have a protumor effect in many or only in a few types of cancer? Recent studies showed that complement activation enhances growth of ovarian, cervical, and non–small-cell lung cancer and sarcoma, and deposition of complement proteins is detectable in more cancer cell types (Table 1). Which complement pathways are activated in cancer? Evidence supporting activation of classical, lectin, and alternative pathways in cancer exists. It is possible that in different types of cancer different pathways are functional. Therefore, it is more reasonable to target common complement proteins or receptors in antitumor therapies. What are the effects of early (C3a and C5a) versus late (MAC) complement activation end products on cancer cells? If higher concentrations of C3a or C5a promote, and denser MAC deposit reduces, tumor growth, developing bispecific inhibitory antibodies targeting C5aR (or C3aR) and CD59 simultaneously may increase the potency of the antitumor effect of complement therapy. Complement-dependent cytotoxicity has been considered an important component of the therapeutic benefit of monoclonal antibodies in malignant B cell disorders; however, the effects of complement activation on white blood cell dyscrasia have not been studied. A few reports point to a prognostic significance of expression of complement genes in leukemic blasts (86, 87). More comprehensive studies on the role of complement activation in leukemia and lymphoproliferative disorders might reveal possible therapeutic benefits of anticomplement reagents in these disorders. Eculizumab, a humanized anti-C5 monoclonal antibody, is currently available on the market and is used to treat paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. A single i.v. infusion of eculizumab blocks complement activation in plasma for 2–3 weeks (88, 89), but its potency and half-life in the interstitial tissue are unknown. Eculizumab blocks generation of C5a and MAC, but would not affect synthesis and secretion of complement proteins by cancer cells or C3a generation in the tumor microenvironment. Currently, no ongoing clinical trials are evaluating eculizumab in cancer patients; however, because of the clinical use of this reagent for other indications, we have a relatively clear picture of its side effect profile. Patients on eculizumab are at risk for developing infections with encapsulated microorganisms and should receive meningococcal vaccination before initiation of therapy. Lack of bone marrow suppression with eculizumab is a therapeutic advantage that can be used in designing clinical trials combining this reagent with chemotherapeutic reagents in cancer patients. In a few animal studies, C5aR antagonists, including PMX-53, have been shown to be effective in reducing tumor size in mice (67, 71), but this or similar reagents have not entered into clinical practice yet. Targeting C5aR rather than C5 or C3 might have the potential benefit of leaving opsonization and MAC generation intact. Intact opsonization of bacteria would reduce the risk of infectious complications in individuals undergoing treatment, and the generation of lytic concentrations of MAC might have a tumoricidal effect. On the other hand, targeting C5aR has the disadvantage of leaving other complement effector molecules, such as C3a, uninhibited. A potential advantage of using anticomplement reagents in cancer treatment is that they can be combined with traditional chemotherapies without increasing myelosuppression associated with chemotherapies; and combined with immune checkpoint inhibitors, because they have different targets. While checkpoint inhibitors increase proliferation of cytotoxic T cells, complement inhibitors decrease MDSCs infiltrating into the tumor microenvironment, reduce MDSC-induced T cell suppression, and enhance T cell function. Based on experiences collected with the clinical use of eculizumab, another advantage of complement inhibitors is their relatively few side effects. Any therapeutic use of anticomplement therapies in solid or liquid tumors should be carefully balanced with possible interference of complement inhibition with the efficacy of other antitumor reagents: (a) The outcome of combining anticomplement reagents with monoclonal antibodies (such as cetuximab, rituximab, or trastuzumab) may depend on the importance of complement-dependent cytotoxicity in the function of these antibodies. (b) Chimeric antigen receptor (CAR) T cell therapies depend on in vitro expansion and in vivo proliferation of T cells. Complement inhibition may decrease the proliferation of CAR T cells in vivo and may reduce their efficacy. Conclusions By mediating cell-cell and cell-stroma interactions, complement proteins have several immune and nonimmune functions in both plasma and the extravascular interstitial tissue. Activation of the complement system in the tumor microenvironment enhances tumor growth via different mechanisms. Anticomplement reagents might have a place in the therapeutic armamentarium against cancer and, because of their limited non-myelosuppressive side effects and nonoverlapping pharmacodynamics, could be combined with traditional chemotherapies or immunotherapies. Acknowledgments This work is supported in part by NIH grant CA177909 (to VAK). The author thanks Michael Kroll for his valuable comments. Footnotes Conflict of interest: The author has declared that no conflict of interest exists. Reference information: J Clin Invest. 2017;127(3):780–789. https://doi.org/10.1172/JCI90962. References Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci. 2002;27(2):67–74. View this article via: PubMed CrossRef Google Scholar Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30(2):83–90. View this article via: PubMed CrossRef Google Scholar Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9(10):729–740. View this article via: PubMed Google Scholar Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–1066. View this article via: PubMed CrossRef Google Scholar Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140–1144. View this article via: PubMed CrossRef Google Scholar Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol. 2014;32:433–459. View this article via: PubMed CrossRef Google Scholar Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol. 2003;40(7):423–429. View this article via: PubMed CrossRef Google Scholar Jack DL, Klein NJ, Turner MW. Mannose-binding lectin: targeting the microbial world for complement attack and opsonophagocytosis. Immunol Rev. 2001;180:86–99. View this article via: PubMed CrossRef Google Scholar Frank MM. Complement in the pathophysiology of human disease. N Engl J Med. 1987;316(24):1525–1530. View this article via: PubMed CrossRef Google Scholar Tegla CA, et al. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res. 2011;51(1):45–60. View this article via: PubMed CrossRef Google Scholar Matthews KW, Mueller-Ortiz SL, Wetsel RA. Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol. 2004;40(11):785–793. View this article via: PubMed CrossRef Google Scholar Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34–50. View this article via: PubMed CrossRef Google Scholar Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171(3):715–727. View this article via: PubMed CrossRef Google Scholar Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190(8):3831–3838. View this article via: PubMed CrossRef Google Scholar Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37(2):199–207. View this article via: PubMed CrossRef Google Scholar Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7(1):9–18. View this article via: PubMed CrossRef Google Scholar Strainic MG, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28(3):425–435. View this article via: PubMed CrossRef Google Scholar Raedler H, Yang M, Lalli PN, Medof ME, Heeger PS. Primed CD8(+) T-cell responses to allogeneic endothelial cells are controlled by local complement activation. Am J Transplant. 2009;9(8):1784–1795. View this article via: PubMed CrossRef Google Scholar Peng Q, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood. 2008;111(4):2452–2461. View this article via: PubMed CrossRef Google Scholar Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8(6):582–587. View this article via: PubMed CrossRef Google Scholar Sacks SH. Complement fragments C3a and C5a: the salt and pepper of the immune response. Eur J Immunol. 2010;40(3):668–670. View this article via: PubMed CrossRef Google Scholar Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112(5):1759–1766. View this article via: PubMed CrossRef Google Scholar Jacquier-Sarlin MR, Gabert FM, Villiers MB, Colomb MG. Modulation of antigen processing and presentation by covalently linked complement C3b fragment. Immunology. 1995;84(1):164–170. View this article via: PubMed Google Scholar Kerekes K, Cooper PD, Prechl J, Józsi M, Bajtay Z, Erdei A. Adjuvant effect of gamma-inulin is mediated by C3 fragments deposited on antigen-presenting cells. J Leukoc Biol. 2001;69(1):69–74. View this article via: PubMed Google Scholar Zhou W, Medof ME, Heeger PS, Sacks S. Graft-derived complement as a mediator of transplant injury. Curr Opin Immunol. 2007;19(5):569–576. View this article via: PubMed CrossRef Google Scholar Carmona-Fontaine C, et al. Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev Cell. 2011;21(6):1026–1037. View this article via: PubMed CrossRef Google Scholar Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. View this article via: PubMed CrossRef Google Scholar Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. View this article via: PubMed CrossRef Google Scholar Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16(12):1773–1782. View this article via: PubMed CrossRef Google Scholar Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183. View this article via: PubMed CrossRef Google Scholar Strey CW, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198(6):913–923. View this article via: PubMed CrossRef Google Scholar Mastellos D, Lambris JD. Complement: more than a ‘guard’ against invading pathogens? Trends Immunol. 2002;23(10):485–491. View this article via: PubMed CrossRef Google Scholar Del Rio-Tsonis K, Tsonis PA, Zarkadis IK, Tsagas AG, Lambris JD. Expression of the third component of complement, C3, in regenerating limb blastema cells of urodeles. J Immunol. 1998;161(12):6819–6824. View this article via: PubMed Google Scholar Reca R, et al. Functional receptor for C3a anaphylatoxin is expressed by normal hematopoietic stem/progenitor cells, and C3a enhances their homing-related responses to SDF-1. Blood. 2003;101(10):3784–3793. View this article via: PubMed CrossRef Google Scholar Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. View this article via: PubMed CrossRef Google Scholar Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339(6117):286–291. View this article via: PubMed CrossRef Google Scholar Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. View this article via: PubMed CrossRef Google Scholar Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. View this article via: PubMed CrossRef Google Scholar Bonavita E, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. 2015;160(4):700–714. View this article via: PubMed CrossRef Google Scholar Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol. 2013;25(1):54–64. View this article via: PubMed CrossRef Google Scholar Nitta H, Murakami Y, Wada Y, Eto M, Baba H, Imamura T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. 2014;32(4):1715–1719. View this article via: PubMed Google Scholar Bulla R, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. 2016;7:10346. View this article via: PubMed Google Scholar Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40(2–4):109–123. View this article via: PubMed Google Scholar Gancz D, Fishelson Z. Cancer resistance to complement-dependent cytotoxicity (CDC): Problem-oriented research and development. Mol Immunol. 2009;46(14):2794–2800. View this article via: PubMed CrossRef Google Scholar Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. View this article via: PubMed CrossRef Google Scholar Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797. View this article via: PubMed CrossRef Google Scholar Gorter A, Meri S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today. 1999;20(12):576–582. View this article via: PubMed CrossRef Google Scholar Nishioka K, Kawamura K, Hirayama T, Kawashima T, Shimada K. The complement system in tumor immunity: significance of elevated levels of complement in tumor bearing hosts. Ann N Y Acad Sci. 1976;276:303–315. View this article via: PubMed CrossRef Google Scholar Chow MT, Moller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012;22(1):23–32. View this article via: PubMed CrossRef Google Scholar Markiewski MM, Lambris JD. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. 2009;30(6):286–292. View this article via: PubMed CrossRef Google Scholar Taylor RP, Lindorfer MA. Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti-tumor monoclonal antibodies. Semin Immunol. 2016;28(3):309–316. View this article via: PubMed CrossRef Google Scholar Di Gaetano N, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171(3):1581–1587. View this article via: PubMed CrossRef Google Scholar Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111(3):1456–1463. Wang SY, et al. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood. 2009;114(26):5322–5330. Pedersen IM, Buhl AM, Klausen P, Geisler CH, Jurlander J. The chimeric anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic leukemia cells through a p38 mitogen activated protein-kinase-dependent mechanism. Blood. 2002;99(4):1314–1319. Kheirallah S, et al. Rituximab inhibits B-cell receptor signaling. Blood. 2010;115(5):985–994. Prang N, et al. Cellular and complement-dependent cytotoxicity of Ep-CAM-specific monoclonal antibody MT201 against breast cancer cell lines. Br J Cancer. 2005;92(2):342–349. View this article via: PubMed Google Scholar Ward PA. Functions of C5a receptors. J Mol Med (Berl). 2009;87(4):375–378. View this article via: PubMed CrossRef Google Scholar Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002;61(2):456–463. View this article via: PubMed CrossRef Google Scholar Kurihara R, et al. C5a promotes migration, proliferation, and vessel formation in endothelial cells. Inflamm Res. 2010;59(8):659–666. View this article via: PubMed CrossRef Google Scholar Cao Q, McIsaac SM, Stadnyk AW. Human colonic epithelial cells detect and respond to C5a via apically expressed C5aR through the ERK pathway. Am J Physiol Cell Physiol. 2012;302(12):C1731–C1740. View this article via: PubMed CrossRef Google Scholar Zhou X, et al. Complement 3 activates the renal renin-angiotensin system by induction of epithelial-to-mesenchymal transition of the nephrotubulus in mice. Am J Physiol Renal Physiol. 2013;305(7):F957–F967. View this article via: PubMed CrossRef Google Scholar Cho MS, et al. Complement component 3 is regulated by TWIST1 and mediates epithelial-mesenchymal transition. J Immunol. 2016;196(3):1412–1418. View this article via: PubMed CrossRef Google Scholar Niculescu F, Badea T, Rus H. Sublytic C5b-9 induces proliferation of human aortic smooth muscle cells: role of mitogen activated protein kinase and phosphatidylinositol 3-kinase. Atherosclerosis. 1999;142(1):47–56. View this article via: PubMed CrossRef Google Scholar Soane L, Cho HJ, Niculescu F, Rus H, Shin ML. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J Immunol. 2001;167(4):2305–2311. View this article via: PubMed CrossRef Google Scholar Kraus S, Seger R, Fishelson Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin Exp Immunol. 2001;123(3):366–374. View this article via: PubMed CrossRef Google Scholar Markiewski MM, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225–1235. View this article via: PubMed CrossRef Google Scholar Peranzoni E, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22(2):238–244. View this article via: PubMed CrossRef Google Scholar Ochando JC, Chen SH. Myeloid-derived suppressor cells in transplantation and cancer. Immunol Res. 2012;54(1-3):275–285. View this article via: PubMed CrossRef Google Scholar Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–999. View this article via: PubMed CrossRef Google Scholar Nunez-Cruz S, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. 2012;14(11):994–1004. View this article via: PubMed CrossRef Google Scholar Connolly DC, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63(6):1389–1397. View this article via: PubMed Google Scholar Cho MS, et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014;6(6):1085–1095. View this article via: PubMed CrossRef Google Scholar Wang Y, et al. et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. 2016;6(9):1022–1035. View this article via: PubMed CrossRef Google Scholar Mamidi S, Höne S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology. 2017;222(1):45–54. View this article via: PubMed CrossRef Google Scholar Shi XX, Zhang B, Zang JL, Wang GY, Gao MH. CD59 silencing via retrovirus-mediated RNA interference enhanced complement-mediated cell damage in ovary cancer. Cell Mol Immunol. 2009;6(1):61–66. View this article via: PubMed CrossRef Google Scholar Donin N, Jurianz K, Ziporen L, Schultz S, Kirschfink M, Fishelson Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. 2003;131(2):254–263. View this article via: PubMed CrossRef Google Scholar Lovisa S, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21(9):998–1009. View this article via: PubMed CrossRef Google Scholar Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. View this article via: JCI PubMed CrossRef Google Scholar Wan J, Zhou X, Cui J, Zou Z, Xu Y, You D. Role of complement 3 in TNF-α-induced mesenchymal transition of renal tubular epithelial cells in vitro. Mol Biotechnol. 2013;54(1):92–100. View this article via: PubMed CrossRef Google Scholar Zhou M, Ma H, Lin H, Qin J. Induction of epithelial-to-mesenchymal transition in proximal tubular epithelial cells on microfluidic devices. Biomaterials. 2014;35(5):1390–1401. View this article via: PubMed CrossRef Google Scholar Goswami MT, et al. Regulation of complement-dependent cytotoxicity by TGF-β-induced epithelial-mesenchymal transition. Oncogene. 2016;35(15):1888–1898. View this article via: PubMed CrossRef Google Scholar Macor P, Tedesco F. Complement as effector system in cancer immunotherapy. Immunol Lett. 2007;111(1):6–13. View this article via: PubMed CrossRef Google Scholar Zhao WP, Zhu B, Duan YZ, Chen ZT. Neutralization of complement regulatory proteins CD55 and CD59 augments therapeutic effect of herceptin against lung carcinoma cells. Oncol Rep. 2009;21(6):1405–1411. Hu W, et al. Human CD59 inhibitor sensitizes rituximab-resistant lymphoma cells to complement-mediated cytolysis. Cancer Res. 2011;71(6):2298–2307. View this article via: PubMed CrossRef Google Scholar Laverdière I, et al. Complement cascade gene expression defines novel prognostic subgroups of acute myeloid leukemia. Exp Hematol. 2016;44(11):1039–1043.e10. View this article via: PubMed CrossRef Google Scholar Abdelbaset-Ismail A, et al. Activation of the complement cascade enhances motility of leukemic cells by downregulating expression of HO-1. [published online ahead of print August 26, 2016]. Leukemia. https://doi.org/10.1038/leu.2016.198. View this article via: PubMed CrossRef Google Scholar Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V, , French Study Group for aHUS/C3G. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8(11):643–657. View this article via: PubMed CrossRef Google Scholar Cugno M, et al. Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Thromb Haemost. 2014;12(9):1440–1448. View this article via: PubMed CrossRef Google Scholar Canales NA, et al. A1BG and C3 are overexpressed in patients with cervical intraepithelial neoplasia III. Oncol Lett. 2014;8(2):939–947. View this article via: PubMed Google Scholar Lin K, et al. Complement component 3 is a prognostic factor of non–small cell lung cancer. Mol Med Rep. 2014;10(2):811–817. View this article via: PubMed Google Scholar Bouwens TA, Trouw LA, Veerhuis R, Dirven CM, Lamfers ML, Al-Khawaja H. Complement activation in Glioblastoma multiforme pathophysiology: evidence from serum levels and presence of complement activation products in tumor tissue. J Neuroimmunol. 2015;278:271–276. View this article via: PubMed CrossRef Google Scholar Nabizadeh JA, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J Immunol. 2016;196(11):4783–4792. View this article via: PubMed CrossRef Google Scholar Habermann JK, et al. Increased serum levels of complement C3a anaphylatoxin indicate the presence of colorectal tumors. Gastroenterology. 2006;131(4):1020–1029; quiz 1284. View this article via: PubMed Google Scholar Bjørge L, et al. Ascitic complement system in ovarian cancer. Br J Cancer. 2005;92(5):895–905. View this article via: PubMed CrossRef Google Scholar Corrales L, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189(9):4674–4683. View this article via: PubMed CrossRef Google Scholar Nitta H, et al. Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88). Clin Cancer Res. 2013;19(8):2004–2013. View this article via: PubMed CrossRef Google Scholar Baatrup G, Qvist N, Junker A, Larsen KE, Zimmermann-Nielsen C. Activity and activation of the complement system in patients being operated on for cancer of the colon. Eur J Surg. 1994;160(9):503–510. View this article via: PubMed Google Scholar Ajona D, et al. Complement activation product C4d in oral and oropharyngeal squamous cell carcinoma. Oral Dis. 2015;21(7):899–904. View this article via: PubMed CrossRef Google Scholar Chen J, Yang WJ, Sun HJ, Yang X
|
|
Scooped by
Gilbert C FAURE
October 2, 2017 2:35 AM
|
Aging is the greatest risk factor for developing chronic diseases. Inflamm-aging, the age-related increase in low-grade chronic inflammation, may be a common link in age-related diseases. This review summarizes recent published data on potential cellular and molecular mechanisms of the age-related increase in inflammation, and how these contribute to decreased humoral immune responses in aged mice and humans. Briefly, we cover how aging and related inflammation decrease antibody responses in mice and humans, and how obesity contributes to the mechanisms for aging through increased inflammation. We also report data in the literature showing adipose tissue infiltration with immune cells and how these cells are recruited and contribute to local and systemic inflammation. We show that several types of immune cells infiltrate the adipose tissue and these include macrophages, neutrophils, NK cells, innate lymphoid cells (ILCs), eosinophils, T cells, B1 and B2 cells. Our main focus is how the adipose tissue affects immune responses, in particular B cell responses and antibody production. The role of leptin in generating inflammation and decreased B cell responses is also discussed. We report data published by us and by other groups showing that the adipose tissue generates pro-inflammatory B cell subsets which induce pro-inflammatory T cells, promote insulin resistance and secrete pathogenic autoimmune antibodies.
|
|
Scooped by
Gilbert C FAURE
December 17, 2015 4:42 PM
|
Eosinophils perform numerous tasks. They are involved in inflammatory reactions associated with innate immune defence against parasitic infections and are also involved in pathological processes in response to allergens.
|
|
Scooped by
Gilbert C FAURE
May 30, 2015 4:45 AM
|
Tumour immunology Eosinophils — T cells' little helpers
Nature.com
Eosinophilia is frequently observed in cancer and eosinophils are attracted to tumours but it is still unknown whether they play an active part in tumour rejection.
|