Your new post is loading...
Your new post is loading...

|
Scooped by
I2BC Paris-Saclay
Today, 7:28 AM
|
The I2BC crystallization platform is evolving into a crystallography platform, offering new services
The crystallization platform has evolved into a crystallography platform and now offers new services (including protein structure determination and/or analysis) as well as scientific support for clients in structural biology projects. Protein crystallography is the main approach for characterizing the 3D structure of proteins associated with drug candidates and for determining the molecular basis of complexes between soluble or membrane proteins and their partners (DNA, RNA, other proteins, peptides, lipids, cofactors, various ligands including metals). Initially, the crystallization platform was dedicated to automated screening, analysis, and optimization of crystallization conditions for soluble and membrane macromolecules. Now, the crystallography platform provides extended services, including protein structure determination and/or analysis. The successive steps, starting from a purified protein sample of interest, include crystal formation, diffraction image collection at the Synchrotron Soleil, electron density map calculation, atomic model refinement of the protein or complex of interest, and structural analysis. Emphasis is placed on obtaining the most accurate models for the most successful structural analysis. Additionally, an AlphaFold service, including predicted model analysis, is available, relying on services established at I2BC by the BIOI2 integrative bioinformatics platform. Routine upstream steps before obtaining a protein crystal include obtaining an optimized synthetic gene for expression in prokaryotic hosts, expression, and purification of the recombinant protein of interest. The platform can advise clients on these various steps, and collaboration or service provision can be considered depending on project complexity. The crystallography platform is open to all scientists, from beginners to experienced crystallographers. It is important to note that the quality of the protein sample is a crucial criterion. The sample must be pure and verified on a denaturing acrylamide gel before crystallization. Do not hesitate to contact us. Contact: cristallographie@i2bc.paris-saclay.fr
More information: https://www.i2bc.paris-saclay.fr/structural-biology/crystallography/
|
|
Scooped by
I2BC Paris-Saclay
March 4, 2:20 AM
|
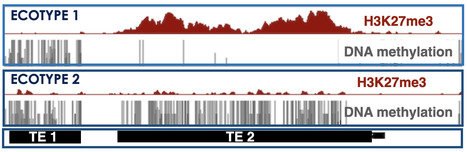
Alternative silencing states of transposable elements in Arabidopsis associated with H3K27me3
This study sheds light on an alternative mode of Transposable Element silencing associated with H3K27me3 instead of DNA methylation in flowering plants; it also indicates dynamic switching between the two epigenetic marks at the species level, a new paradigm that might extend to other multicellular eukaryotes. The DNA/H3K9 methylation and Polycomb-group proteins (PcG)-H3K27me3 silencing pathways have long been considered mutually exclusive and specific to transposable elements (TEs) and genes, respectively in mammals, plants, and fungi. However, H3K27me3 can be recruited to many TEs in the absence of DNA/H3K9 methylation machinery and sometimes also co-occur with DNA methylation.
In this study, the team of Angélique Déléris showed that TEs can also be solely targeted and silenced by H3K27me3 in wild-type Arabidopsis plants. These H3K27me3-marked TEs not only comprise degenerated relics but also seemingly intact copies that display the epigenetic features of responsive PcG target genes as well as an active H3K27me3 regulation. The authors also showed that H3K27me3 can be deposited on newly inserted transgenic TE sequences in a TE-specific manner indicating that silencing is determined in cis. Finally, a comparison of Arabidopsis natural accessions reveals the existence of a category of TEs—which we refer to as “bifrons”—that are marked by DNA methylation or H3K27me3 depending on the accession. This variation can be linked to intrinsic TE features and to trans-acting factors and reveals a change in epigenetic status across the TE lifespan.
Overall, this study shedded light on an alternative mode of TE silencing associated with H3K27me3 instead of DNA methylation in flowering plants. It also suggests dynamic switching between the two epigenetic marks at the species level, a new paradigm that might extend to other multicellular eukaryotes. More Information: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03466-6 Contact: Angélique Déléris angelique.deleris@i2bc.apris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
February 17, 4:00 AM
|
ChemPhysBio2025 : Registrations Now Open!
We’re excited to announce that registrations are now open for the Second Interdisciplinary Summer School: ChemPhysBio2025 – Imaging Biology: Interdisciplinary Tools and Techniques to Unravel Cellular Dynamics – taking place June 16–20, 2025, at Université Paris-Saclay! This unique event bridges chemistry, physics, and biology, offering participants the opportunity to explore advanced photonic imaging techniques for unraveling cellular dynamics. We’re also thrilled to welcome two distinguished guest lecturers: - Julie Karpenko (Laboratory for Therapeutic Innovation, University of Strasbourg)
- Balázs Enyedi (Department of Physiology, Semmelweis University, Hungary)
What to expect? - Inspiring lectures and research seminars
- Interactive interdisciplinary workshops
- Hands-on experience in cutting-edge research labs
Who should apply?
Master’s students, PhD candidates, postdocs, and junior researchers who are newcomers to the interdisciplinary field and eager to deepen their knowledge, expand their skill sets, and build meaningful collaborations. With only 20 spots available, don’t miss your chance to join us!
Apply now: https://chemphysbio2025.sciencesconf.org/
|
|
Scooped by
I2BC Paris-Saclay
February 11, 8:48 AM
|
Complete description of the biosynthetic process of [2Fe-2S] clusters
Iron-sulfur clusters are essential metallocofactors providing catalytic activities to a multitude of enzymes and proteins. Here, we report a comprehensive picture of their assembly mechanism, showing that it relies on the formation of [1Fe-1S] precursors fused into [2Fe-2S] clusters upon dimerization of the scaffold protein. Iron-sulfur (Fe-S) clusters are ubiquitous metallocofactors constituting the active site of a multitude of enzymes and proteins involved in electron transfer, catalysis, sulfur donation and signalling. They are made of iron and sulfide ions assembled into diverse structures. The [2Fe-2S] and [4Fe-4S] clusters are the most common forms in organisms. They are synthesized by multi-protein machineries which have remained highly conserved during evolution. The iron-sulfur cluster (ISC) assembly machinery present in eukaryotes and prokaryotes synthesizes [2Fe-2S] clusters, which serve as building blocks for the assembly of [4Fe-4S] clusters. The core ISC machinery assembles [2Fe-2S] clusters on the scaffold protein IscU, which requires iron provided by an unknown source, sulfur provided in the form of cysteine bound persulfides (Cys-SSH) by the cysteine desulfurase IscS, and electrons provided by the ferredoxin–ferredoxin reductase complex Fdx-FdxR from NADPH. Then, specialized chaperones transfer [2Fe-2S] clusters to recipient acceptors. Despite previous studies on the core assembly machinery, the mechanistic details of the [2Fe-2S] cluster assembly process have remained poorly understood due to the experimental difficulties in trapping the relevant intermediates.
The team Biochemistry of Metalloproteins and Associated Diseases at the Institute of Integrative Biology of the Cell in Gif-Sur-Yvette (I2BC, UMR 9198, CNRS – CEA - Paris-Saclay University) managed to dissect this process step by step and to isolate several key intermediates using a functional reconstitution of the Escherichia coli ISC machinery. They used a combination of biochemical techniques to trap these intermediates: anaerobic reconstitution, persulfide detection assays, kinetics, UV-visible, circular dichroism, and to characterize them by spectroscopic methods: electron paramagnetic spectroscopy (EPR), nuclear magnetic resonance (NMR) and native mass spectrometry (nMS) in collaboration with teams at Aix-Marseille University (BIP), Gif-Sur-Yvette (ICSN) and Strasbourg (IPHC). They show that the assembly of [2Fe-2S] clusters is initiated by iron binding to IscU, which triggers persulfide insertion by IscS in the vicinity of the iron-binding site of IscU upon the formation of a complex between IscU and IscS. The persulfide in IscU binds to the iron center and is cleaved into sulfide by the Fdx-FdxR complex, which leads to the formation of a Fe-SH intermediate, referred to as the [1Fe-1S] precursor. Then, IscU dissociates from IscS, dimerizes and generates a bridging [2Fe-2S] cluster by fusion of two [1Fe-1S] precursors. The IscU dimer ultimately dissociates into a monomer, ready to transfer its [2Fe-2S] cluster to acceptors. The data also indicate that the bridging cluster is initially in the super-reduced state [2Fe-2S]0 and releases two electrons to the ferredoxin enzyme, thereby leading to an oxidised [2Fe-2S]2+ state as the final product.
These data provide a comprehensive description of mechanism of [2Fe-2S] clusters assembly by the bacterial ISC machinery, highlighting the formation of key intermediates through a tightly concerted process. This stepwise dissection further supports findings in eukaryotes, including iron loading, persulfidation and dimerization of IscU, which point to an evolutionary conservation of the assembly process. More information: https://www.nature.com/articles/s41589-024-01818-8 Contact: Benoit D'Autréaux benoit.dautreaux@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
February 10, 8:09 AM
|
Tutorial videos for ChimeraX
|
|
Scooped by
I2BC Paris-Saclay
January 21, 8:18 AM
|
The Biology and Physics of Prokaryotic Chromosomes VII
The Biology and Physics of Prokaryotic Chromosome is an international scientific conference, bringing together experimentalists and theoreticians to discuss discoveries and challenges around building an integrated model of prokaryotic chromosome structure. The conference focuses on mechanisms of chromosome organisation and structure, chromosome replication, transcription, and segregation, investigated at multiple scale lengths, using both experimental and theoretical approaches. A key goal is to provide a platform for inter-disciplinary work, to foster new collaboration and develop new ideas. New approaches and technologies have revolutionised the field of prokaryotic chromosome biology in the last fifteen years. For example, super-resolution microscopy is now being used to visualize previously unresolved structure, genomic tools have enabled the capture of new types of proximity information on a genomic-scale, and polymer physics models allow researchers to tackle questions that go far beyond the traditional biological description of chromosome structure. Join us in York for an unforgettable scientific meeting - this is an opportunity you won’t want to miss. At least half of the oral presentation slots are expected to be selected from submitted abstracts and we will prioritise early career researchers. Programme Coordinators:
David Grainger, University of Birmingham
Daniela Barillà, University of York
Remus Dame, Leiden University
Virginia Lioy, Institute for Integrative Biology of the Cell More information: bit.ly/bio-prokaryotic-VII Contact: Virginia LIOY virginia.lioy@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
January 16, 7:23 AM
|
Newcomer at the BIOI2 bioinformatics platform
Baptiste Roelens joined the BIOI2 platform last month, bringing expertise in machine learning for biological data analysis. His contributions will expand the platform's service offerings and support the I2BC imaging community. The integrative BIOinformatics platform (BIOI2) welcomes Baptiste Roelens as a new engineer. Baptiste's background combines theoretical computer science and life sciences, with a foundation from Ecole Polytechnique. He further honed his skills at the Institut Curie, where he earned his PhD. His doctoral research focused on developing quantitative approaches to analyze the role of the histone chaperone CAF-1 in maintaining higher-order chromatin structure during meiotic prophase in the fruit fly. Continuing his research journey, Baptiste joined Stanford University to develop bioimage analysis pipelines, further exploring the mechanisms regulating chromosome organization and cell-cycle progression in C. elegans germline. Upon returning to France, he contributed to the biotech company Viroxis, developing cutting-edge deep-learning approaches for protein design. With experience spanning both wet and dry labs, Baptiste will be a great asset to the BIOI2 platform and is ideally placed to collaborate with the I2BC’s imaging community, in particular the Imagerie-Gif platform (https://www.i2bc.paris-saclay.fr/bioimaging/), to develop pipelines that streamline the processing and analysis of complex imaging data. BIOI2 provides access to bioinformatics resources and activities developed and of use at the I2BC, organises focused training session on bioinformatics tools, and is an official contributing plateform of the nationwide French Institute of Bioinformatics (IFB). More information: https://bioi2.i2bc.paris-saclay.fr Contact: contact-bioi2@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
December 16, 2024 4:49 AM
|
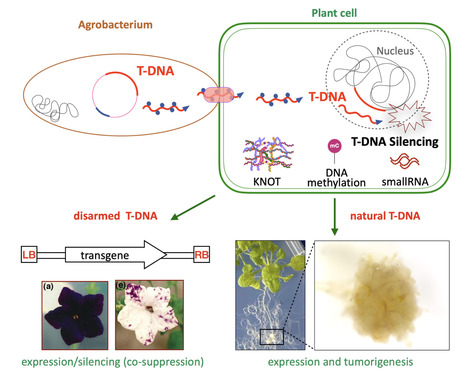
Epigenetic control of T-DNA during transgenesis and pathogenesis
T-DNAs are mobile elements transferred from pathogenic Agrobacterium to plants that reprogram host cells into hairy roots or tumors, and which are used as disarmed forms to deliver transgenes in plants. Here we review the mechanisms that silence the expression of T-DNAs in transgenic plants as well as during pathogenesis. Mobile elements known as T-DNAs are transferred from pathogenic Agrobacterium to plants and reprogram the host cell to form hairy roots or tumors. Disarmed nononcogenic T-DNAs are extensively used to deliver transgenes in plant genetic engineering. Such T-DNAs were the first known targets of RNA silencing mechanisms, which detect foreign RNA in plant cells and produce small RNAs that induce transcript degradation. These T-DNAs can also be transcriptionally silenced by the deposition of epigenetic marks such as DNA methylation and the dimethylation of lysine 9 (H3K9me2) in plants. Here, we review the targeting and the roles of RNA silencing and DNA methylation on T-DNAs in transgenic plants as well as during pathogenesis. In addition, we discuss the crosstalk between T-DNAs and genome-wide changes in DNA methylation during pathogenesis. We also cover recently discovered regulatory phenomena, such as T-DNA suppression and RNA silencing-independent and epigenetic-independent mechanisms that can silence T-DNAs. Finally, we discuss the implications of findings on T-DNA silencing for the improvement of plant genetic engineering. More information: https://academic-oup-com.insb.bib.cnrs.fr/plphys/advance-article/doi/10.1093/plphys/kiae583/7876130 Contact: Angélique DELERIS angelique.deleris@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
December 4, 2024 10:38 AM
|
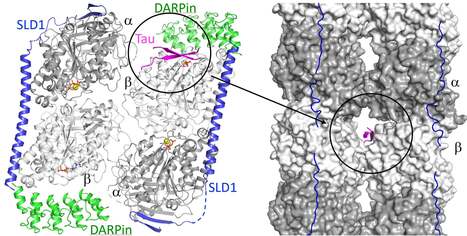
The structure of a Tau fragment bound to tubulin prompts new hypotheses on Tau mechanism and oligomerization
We have identified a novel tubulin surface targeted by the Tau protein. From these results, we formulate new hypotheses on the regulation of microtubules by Tau and on Tau oligomerization. Tau is a protein involved in the regulation of axonal microtubules in neurons. In pathological conditions, it forms filamentous aggregates which are molecular markers of the Alzheimer’s disease and of related neurodegenerative disorders collectively known as tauopathies. Structures of Tau in fibrils or bound to the microtubule have been reported. We have determined the structure of a Tau construct comprising the PHF6 motif, an hexapeptide involved in Tau aggregation, as a complex with tubulin. This Tau fragment binds as a dimer to a new site which, when transposed to the microtubule, would correspond to a pore between protofilaments. These results raise new hypotheses on Tau-induced microtubule assembly and stabilization and on Tau oligomerization. They reconcile apparently contradictory data from the literature, in particular concerning the coexistence of different binding modes of Tau to the microtubule. More information : https://academic.oup.com/pnasnexus/article/3/11/pgae487/7850850 Contact : Benoît GIGANT benoit.gigant@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
November 26, 2024 2:28 AM
|
Perylene-derivative singlet exciton fission in water solution
Ultrafast formation of triplet statates by singlet fission in water solution. We provide evidence that singlet fission—a process where one excited state splits into two—can occur in water-soluble compounds. Specifically, we studied perylene-3,4,9,10-tetracarboxylate, a molecule that forms transient disordered dimers. Using a combination of advanced techniques like time-resolved absorption and fluorescence spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and theoretical modeling, we were able to detect the key features of singlet fission and map out the behavior of the molecular assemblies. Our results show that the constant structural changes within these dimers play a crucial role in steering the process toward either singlet fission or charge separation. The quantum efficiency of producing triplet states is over 100%, with these states lasting for nanoseconds—highlighting how disordered systems can drive highly efficient photophysical processes. More information: https://pubs.rsc.org/en/content/articlehtml/2024/sc/d4sc04732j Contact: Manuel LLANSOLA <manuel.llansola@I2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
November 26, 2024 2:23 AM
|
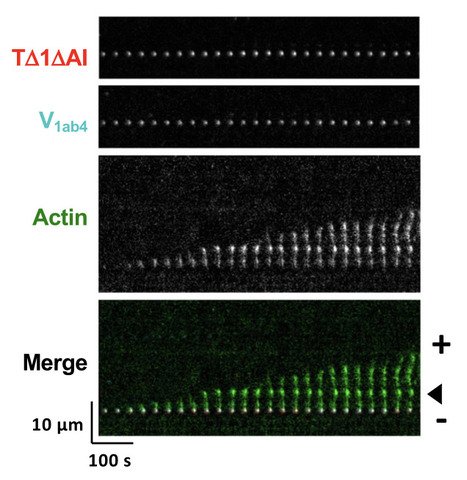
Talin and vinculin combine their activities totrigger actin assembly
In cells, focal adhesions (FAs) strengthen their connection with the actin cytoskeleton to resist force. By combining biochemistry and cell biology, the authors show that the proteins talin and vinculin control actin assembly, thus reinforcing the anchoring of actin to FAs. Focal adhesions (FAs) strengthen their link with the actin cytoskeleton to resistforce. Talin-vinculin association could reinforce actin anchoring to FAs bycontrolling actin polymerization. However, the actin polymerization activity ofthe talin-vinculin complex is not known because it requires the reconstitutionof the mechanical and biochemical activation steps that control the associa-tion of talin and vinculin. By combining kinetic and binding assays with singleactinfilament observations in TIRF microscopy, we show that the associationof talin and vinculin mutants, mimicking mechanically stretched talin andactivated vinculin, triggers a sequential mechanism in whichfilaments arenucleated, capped and released to elongate. In agreement with these obser-vations, FRAP experiments in cells co-expressing the same constitutivemutants of talin and vinculin revealed accelerated growth of stressfibers. Ourfindings suggest a versatile mechanism for the regulation of actin assembly inFAs subjected to various combinations of biochemical and mechanical cues. More information: https://www.nature.com/articles/s41467-024-53859-1 Contact: Christophe LE CLAINCHE <christophe.leclainche@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
November 19, 2024 2:27 AM
|
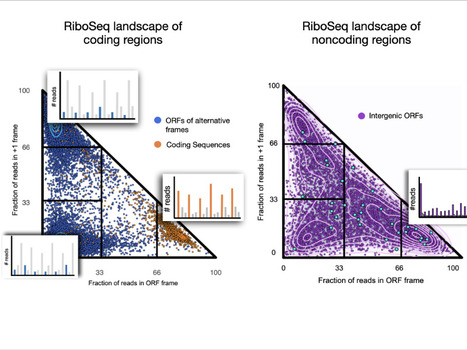
The ribosome profiling landscape of yeast
We explored the ribosome profiling landscape of yeast and showed that noncoding regions are associated with a wide diversity of translation signals among which some led to detectable protein products. Pervasive translation is a widespread phenomenon that plays a critical
role in the emergence of novel microproteins, but the diversity of translation patterns contributing to their generation remains unclear. Based on 54 ribosome profiling (Ribo-Seq) datasets, we investigated the yeast Ribo-Seq landscape using a representation framework that allows the comprehensive inventory and classification of the entire diversity of Ribo-Seq signals, including non-canonical ones. We show that if coding regions occupy specific areas of the Ribo-Seq landscape, noncoding regions encompass a wide diversity of Ribo-Seq signals and, conversely, populate the entire landscape. Our results show that pervasive translation can, nevertheless, be associated with high specificity, with 1055 noncoding ORFs exhibiting canonical Ribo-Seq signals. Using mass spectrometry under standard conditions or proteasome inhibition with an in-house analysis protocol, we report 239 microproteins originating from noncoding ORFs that display canonical but also non-canonical Ribo-Seq signals. Each condition yields dozens of additional microprotein candidates with comparable translation properties, suggesting a larger population of volatile microproteins that are challenging to detect. Our findings suggest that non-canonical translation signals may harbor valuable information and underscore the significance of considering them in proteogenomic studies. Finally, we show that the translation outcome of a noncoding ORF is primarily determined by the initiating codon and the codon distribution in its two alternative frames, rather than features indicative of functionality. Our results enable us to propose a topology of a species’ Ribo-Seq landscape, opening the way to comparative analyses of this translation landscape under different conditions. More information: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03403-7 Contact: Anne Lopes anne.lopes@i2bc.paris-saclay.fr
- Are you interested in Parasitology? or - Would you like to discover a field of research at the interface between Biology and Chemistry? or - Would you like to discover new research methods or study models? Come and attend the Workshop: Practical information: - Date: 11/27/2024
- Times: 9.15 a.m. to 12.15 p.m. (detailed program above)
- Address: Bâtiment Henri Moissan, 17, avenue des Sciences, 91400 ORSAY, France
- Room: 2000 (HM1 building)
- Program and theme (details attached): 3 parasitology specialists come to talk about their latest research findings on malaria and leishmaniasis. Come and listen to their lectures and have lunch with them.
- Registration (deadline 11/20/2024): registration is free but compulsory by clicking on this link
- Module validation: This seminar could validate a doctoral school module for doctoral students.
Via Life Sciences UPSaclay
|
|
|
Scooped by
I2BC Paris-Saclay
March 11, 4:05 PM
|
Borders in our chromosomes shape the neighboring domains
Researchers at the I2BC and the Ecole Normale Supérieure found that the borders that create separation between functional domains in our chromosomes have an unexpectedly large influence on those domains themselves. Chromosomes in humans and other mammals are subdivided into “functional neighborhoods” that guide the fidelity of biological processes, including gene regulation and DNA repair. Perturbations of these neighborhoods, resulting in the fusion of adjacent domains, can lead to a variety of diseases, including cancer and developmental disorders.
In a study published in PNAS (Proceedings of the National Academy of Sciences of the USA), scientists from the Noordermeer group at the I2BC, together with their colleagues at the Ecole Normale Supérieure in Paris, report how chromosomal border elements influence the organization of the adjacent functional neighborhoods. By combining genomics-based analysis and biophysical simulations of chromosome structure, they find that the size of these borders is highly diverse, from simple “points” on the chromosome to highly extended zones of transition between neighborhoods. Computer simulations of chromosome behavior of different types of borders in-between revealed an unexpected and far-reaching impact of the borders on their neighboring domains. Rather than creating a static separation, the borders will actively influence the degree of neighborhood mixing due to a previously unrecognized mechanism whereby the borders reel-in the adjacent neighborhoods. At narrow borders, this actively promotes mixing and may cause interference between biological activity on both side of the border. Extended borders buffer against this mechanism by adding additional separation. Border structure can thus constitute a new regulatory layer in the genome to fine-tune biological processes. More information: https://www.pnas.org/doi/10.1073/pnas.2413112122 Contact: Daan Noordermeer daan.noordermeer@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
March 4, 1:57 AM
|
Ribosome profiling and immunopeptidomics reveal tens of novel conserved HIV-1 ORF encoding T cell antigens.
The translatome of HIV-1 reveals tens of alternative open reading frames (ARF), encoding conserved viral antigens. In vivo, ARF-derived peptides elicit potent HIV-specific poly-functional T cell responses mediated by both CD4+ and CD8+ T cells. T lymphocytes play a pivotal role in controlling human immunodeficiency virus type 1 (HIV-1) infection. Their activation relies on the recognition of viral peptides, or antigens, presented on the surface of infected cells by major histocompatibility complex (MHC) molecules. It is widely assumed that these antigens arise solely from canonical HIV proteins. Previously, we showed that HIV-specific T lymphocytes can also target peptides derived from HIV alternative reading frames (ARFs) presented by MHC molecules. However, evidence for ARFs in the HIV genome had thus far been indirect. In this new study, using ribosome profiling (RiboSeq), we identified the complete HIV-1 translatome in infected CD4⁺ T cells. This approach systematically identified virally encoded mRNA sequences actively translated in HIV-infected cells. We found that the HIV-1 genome contains over one hundred ARFs located either in the 5ʹ untranslated region (5ʹUTR) of classical viral genes or overlapping canonical HIV open reading frames. Using two complementary methods: (1) detecting T lymphocytes specific to ARF-derived peptides in PBMCs from people living with HIV and (2) directly isolating ARF-derived peptides bound to MHC molecules via mass spectrometry–based immunopeptidomics—we demonstrate that HIV ARFs encode novel viral antigens capable of eliciting broad and potent T-cell responses. Our results expand the range of HIV antigens that could be harnessed for vaccine development and may also reveal the existence of microproteins or pseudogenes in the HIV genome.
These findings stem from the work of two complementary teams at I2BC: one led by Olivier Namy, a specialist in protein translation, and the other led by Arnaud Moris, an expert in the immune response to HIV. This collaboration also involved French research and clinical teams and the University of Tübingen in Germany. More information: https://doi.org/10.1038/s41467-025-56773-2 Contact: Arnaud Moris arnaud.moris@i2bc.paris-saclay.fr & Olivier Namy olivier.namy@i2bc.paris-saclay.fr Image: Sonhita Chakraborty
|
|
Scooped by
I2BC Paris-Saclay
February 12, 8:38 AM
|
The Asgard archaeal origins of Arf family GTPases involved in eukaryotic organelle dynamics
How did the endomembrane system, unique to eukaryotes, emerge from their prokaryotic ancestors? We report that Arf proteins, which are crucial regulators of membrane dynamics in eukaryotes, originated in the archaeal ancestor of eukaryotes, the Asgard Archaea. The evolution of eukaryotes is a fundamental event in the history of life. The closest prokaryotic lineage to eukaryotes, the Asgardarchaeota, encode proteins previously found only in eukaryotes, providing insight into their archaeal ancestor. Eukaryotic cells are characterized by endomembrane organelles, and the Arf family GTPases regulate organelle dynamics by recruiting effector proteins to membranes upon activation. The Arf familyis ubiquitous among eukaryotes, but its origins remain elusive. Here we report a group of prokaryotic GTPases, the ArfRs, which are widely present in Asgardarchaeota. Phylogenetic analyses reveal that eukaryotic Arf family proteins arose from the ArfR group. Expression of representative Asgardarchaeota ArfR proteins in yeast and Xray crystallographic studies show that ArfR GTPases possess the mechanism of membrane binding and structural features unique to Arf family proteins. Our results indicate that Arf family GTPases originated in the archaeal ancestor of eukaryotes, consistent with aspects of the endomembrane system evolving early in eukaryogenesis. More information: https://www.nature.com/articles/s41564-024-01904-6#Abs1 Contact: Julie Ménétrey julie.menetrey@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
February 11, 7:55 AM
|
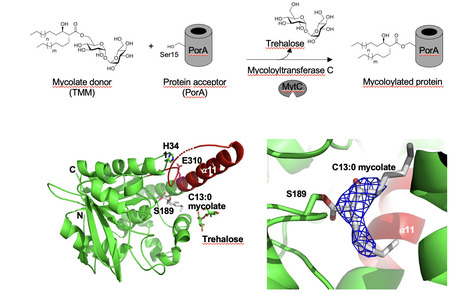
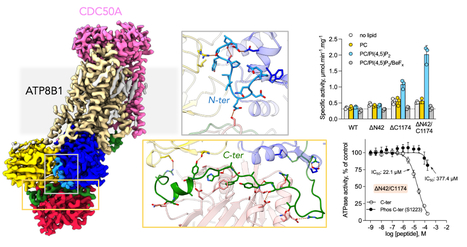
Synthetic mycolates derivatives to decipher protein mycoloylation, a unique post-translational modification in bacteria
In vitro reconstitution of bacterial protein mycoloylation. Protein mycoloylation is a newly characterized post-translational modification (PTM) specifically found in Corynebacteriales, an order of bacteria that includes numerous human pathogens. Their envelope is composed of a unique outer membrane, the so-called mycomembrane made of very-long chain fatty acids, named mycolic acids. Recently, some mycomembrane proteins including PorA have been unambiguously shown to be covalently modified with mycolic acids in the model organism Corynebacterium glutamicum by a mechanism that relies on the mycoloyltransferase MytC. This PTM represents the first example of protein O-acylation in prokaryotes and the first example of protein modification by mycolic acid. Through the design and synthesis of trehalose monomycolate (TMM) analogs, we prove that i) MytC is the mycoloyltransferase directly involved in this PTM, ii) TMM, but not trehalose dimycolate (TDM), is a suitable mycolate donor for PorA mycoloylation, iii) MytC is able to discriminate between an acyl and a mycoloyl chain in vitro unlike other trehalose mycoloyltransferases. We also solved the structure of MytC acyl-enzyme obtained with a soluble short TMM analogs which constitutes the first mycoloyltransferase structure with a covalently linked to an authentic mycolic acid moiety. These data highlight the great conformational flexibility of the active site of MytC during the reaction cycle and pave the way for a better understanding of the catalytic mechanism of all members of the mycoloyltransferase family including the essential Antigen85 enzymes in Mycobacteria. more information: https://doi.org/10.1016/j.jbc.2025.108243 Contact: Florence CONSTANTINESCO-BECKER <florence.constantinesco@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
January 30, 2:50 AM
|
Genetic differentiation in the MAT-proximal region is not sufficient for suppressing recombination in Podospora anserina
A large genomic region in Podospora anserina remains entirely devoid of crossovers, despite being fully colinear. Recombination is advantageous over the long-term, as it allows efficient selection and purging deleterious mutations. Nevertheless, recombination suppression has repeatedly evolved in sex chromosomes. In fungi, sexual compatibility is driven by a locus called mating-type (mat), around which recombination suppression is also often observed.
The evolutionary causes for recombination suppression and the proximal mechanisms preventing crossing overs are poorly understood. Several hypotheses have recently been suggested based on theoretical models, and in particular, that divergence could accumulate neutrally around a sex-determining region and reduce recombination rates, a self-reinforcing process that could foster progressive extension of recombination suppression.
We used the ascomycete fungus Podospora anserina for investigating these questions: a 0.8 Mbp region around its mat locus is non-recombining (called MAT-proximal region), despite being collinear between the two mating types. This fungus is mostly selfing, resulting in highly homozygous individuals, except in the non-recombining region around the mating-type locus that displays differentiation between mating types. Here, we test the hypothesis that sequence divergence alone is responsible for recombination cessation. We replaced one mat idiomorph by the sequence of the other, to obtain compatible strains isogenic in the MAT-proximal region. Crosses showed that recombination was still suppressed in that context, indicating that other proximal mechanisms than inversions or mere sequence divergence are responsible for recombination suppression in this fungus.
This finding suggests that selective mechanisms likely acted for suppressing recombination, or the spread of epigenetic marks, as the neutral model based on mere nucleotide divergence does not seem to hold in P. anserina. More information: https://doi.org/10.1093/g3journal/jkaf015 Contact: Pierre GROGNET pierre.grognet@universite-paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
January 17, 3:17 AM
|
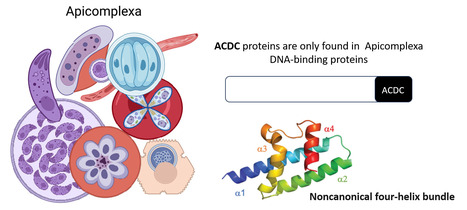
Rocking Science: New ACDC Protein Fold Unveiled
The Apicomplexa-specific ACDC domain adopts a never before seen protein fold. In collaboration with the Nessler team at the I2BC and the Llinás lab at PSU, US, we reveal that the ACDC domain, only found in DNA-binding proteins of the Apicomplexa phylum, grouping several important human pathogens including malaria, has a never before seen protein fold. We also identify potential ligands that may be optimized in the future as protein inhibitors. More information:https://journals.iucr.org/paper?S2059798324012518 Conatct: Joana SANTOS joana.santos@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
December 17, 2024 10:56 AM
|
DciA, the Bacterial Replicative Helicase Loader, promotes LLPS in the presence of ssDNA.
This study shows that DciA, the bacterial replicative helicase loader, promotes condensates in the presence of DNA. This opens the way to the possibility of non-membrane compartments in bacteria. The goal could be to concentrate the players involved in replication and thus facilitate it. The loading of the bacterial replicative helicase DnaB is an essential step for genome replication and depends on the assistance of accessory proteins. Several of these proteins have been identified across the bacterial phyla. DciA is the most common loading protein in bacteria, yet the one whose mechanism is the least understood. We have previously shown that DciA from Vibrio cholerae is composed of a globular domain followed by an unfolded extension and demonstrated its strong affinity for DNA. Here, drawing on the skills of two I2BC facilities, Light microscopy (Imagerie-Gif) and PIM (Structural biology), we characterize the condensates formed by VcDciA upon interaction with a short single-stranded DNA substrate. We demonstrate the fluidity of these condensates using light microscopy and address their network organization through electron microscopy, thereby bridging events to conclude on a liquid-liquid phase separation behavior. Additionally, we observe the recruitment of DnaB in the droplets, concomitant with the release of DciA. We show that the well-known helicase loader DnaC from Escherichia coli is also competent to form these phase-separated condensates in the presence of ssDNA. Our phenomenological data are still preliminary as regards the existence of these condensates in vivo, but open the way for exploring the potential involvement of DciA in the formation of non-membrane compartments within the bacterium to facilitate the assembly of replication players on chromosomal DNA. More information : https://pubmed.ncbi.nlm.nih.gov/39603490/ Contact : Sophie CHERUEL sophie.quevillon-cheruel@i2bc.paris-saclay.fr and Stéphanie MARSIN stephanie.marsin@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
December 9, 2024 2:44 AM
|
Ribosome composition plays a key role in EMT
Ribosome remodeling drives cancer cell transformation: A single protein, RPL36A, triggers EMT. Epithelial-mesenchymal transition (EMT) is a biological process whereby a cell loses its usual characteristics to acquire properties allowing it to move. EMT is involved in embryo formation and tissue repair in adults. However, when altered, EMT contributes to the formation of fibrosis or, in the case of a tumor cell, to the formation of metastases. In cancers, this change is comparable to a cellular metamorphosis that allows cancer cells to become more aggressive and resistant to treatments. EMT is therefore a key player in tumor progression.
EMT is extensively studied at the molecular and cellular levels because a better understanding of this process will enable the development of new therapeutic approaches. However, the translational changes that occur during EMT have so far been very poorly studied.
In this work, we have identified that the overexpression of a single ribosomal protein, called RPL36A, is sufficient to induce EMT. We have also highlighted the various translational changes that occur during EMT.
Our work reveals the importance of ribosome remodeling itself in finely modulating translation during the change in cellular identity that occurs during EMT. More information : https://www.pnas.org/doi/10.1073/pnas.2408114121 ; In French: https://www.insb.cnrs.fr/fr/cnrsinfo/des-modifications-de-la-composition-du-ribosome-accompagnent-les-changements. Contact: Olivier NAMY olivier.namy@i2bc.paris-saclay.fr
Portrait Jeune Chercheur – Christopher Dane Fage, Chercheur en biochimie structurale
Christopher Dane Fage is a structural biochemist who, in January of 2024, joined the Institute for Integrative Biology of the Cell - I2BC (CNRS/CEA/UPSaclay, Gif-sur-Yvette) as a CNRS CRCN to lead the team Structures and functions of hybrid natural product assembly lines. His research seeks to understand and exploit the biosynthesis of medically relevant bacterial metabolites by multi-family enzyme systems. In 2014, Chris earned his PhD from the University of Texas at Austin (USA) in the team of Adrian Keatinge-Clay, where his interest in the structural biochemistry of polyketide synthases (PKSs) crystallized (these multi-domain, multi-module enzymatic assembly lines build complex molecules from simple building blocks through the catalytic chemistry of fatty acid synthases (FASs)). For example, from the spinosyn insecticide pathway, he investigated a Diels-Alderase-like enzyme (catalyzes a reaction widely used in chemical synthesis) and a “dimerization element” (organizes the modular architecture of many PKSs). Subsequently, as a postdoc at Philipps-Universität Marburg (Germany) with Mohamed Marahiel, Chris focused on other types of natural product-forming enzymes, such as non-ribosomal peptide synthetases (NRPSs; these assembly line systems incorporate different building blocks than PKSs using different catalytic domains). For example, he initiated an integrative structure-function study on the inter-subunit “COM” interfaces needed for efficient peptide formation in many NRPSs. He also studied the properties, detection, biosynthesis, and tailoring of the ribosomally synthesized and post-translationally modified lasso peptides, which adopt a remarkably stable lariat knot-like fold. From 2017, Chris joined the teams of Greg Challis and Józef Lewandowski at the University of Warwick (UK). There, he investigated trans-AT PKSs, which employ a wide variety of standalone enzymes in place of their subunit-embedded counterparts, as well as hybrid PKS-NRPS systems, which merge the catalytic chemistry of PKSs and NRPSs – thanks to their peculiarities, these machineries build exceptionally diverse compounds. He also clarified the mode of action of the kalimantacin antibiotics against the human pathogen Staphylococcus aureus. In a brief departure from natural products, Chris worked at the Centre for Medicines Discovery at the University of Oxford (UK) during 2022-2023, where he characterized the structures of three enzymes associated with type 2 diabetes mellitus. He also developed a high-throughput assay for screens against libraries of tens of thousands of inhibitors. Since arriving at the I2BC, Chris has been pursuing structure-guided studies towards the rational reprogramming of hybrid FAS-NRPS-PKS systems, with particular interest in those of the kalimantacin (antibacterial) and bleomycin (anticancer) drugs. “The true delight is in the finding out rather than in the knowing.” - Isaac Asimov -> Contact : christopher.fage@i2bc.paris-saclay.fr
Via Life Sciences UPSaclay
|
|
Scooped by
I2BC Paris-Saclay
November 26, 2024 2:25 AM
|
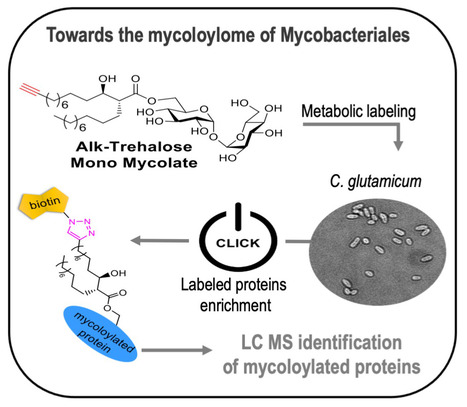
Bioorthogonal monomycolate of trehalose disclosed the O-mycoloylation of mycoloyltransferases and other cell envelope proteins in C. glutamicum
Discovering the mycoloylome of Corynebacterium glutamicum : a new type of lipidated-modified proteins. Protein mycoloylation is a lipidation pathway exclusively observed in Mycobacteriales, an order of bacteria that includes several human pathogens. These bacteria possess a distinctive outer membrane, known as the mycomembrane, composed of very long-chain fatty acids called mycolic acids. It has been demonstrated that a few mycomembrane proteins undergo covalent modification with mycolic acids in the model organism Corynebacterium glutamicum, through the action of mycoloyltransferase MytC. This PTM represents the first example of protein O-acylation in prokaryotes and also the first example of protein modification by mycolic acid. Here, we have developed a unique bioorthogonal mycolate donor featuring the natural mycolic acid pattern, enabling direct unambiguous transfer of the lipid moiety to its acceptors and efficient metabolic labeling and enrichment of MytC protein substrates. Mass spectrometry analysis of the labeled proteins and comparative proteomic analysis of the cell envelope proteome between wild-type and ∆mytC strains identified an unbiased list of 21 proteins likely mycoloylated in the cell. The robustness of our approach is demonstrated by the successful biological validation of mycoloylation in 6 random candidate proteins within wild-type cells, revealing the characteristic profile of proteins modified with natural mycolates. These findings provide interesting insights into the significance of this new lipidation pathway and pave the way for understanding their function, especially concerning the mycoloyltransferase family that includes the essential Antigen85 enzymes in Mycobacteria. More information: https://doi.org/10.1021/acschembio.4c00502. Contact: Cécile LABARRE <cecile.labarre@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
November 19, 2024 2:50 AM
|
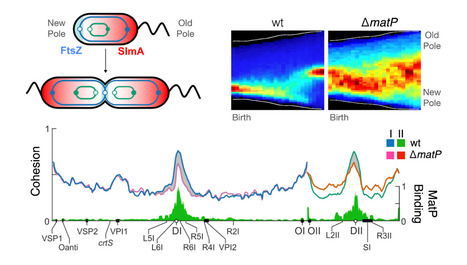
MatP local enrichment delays segregation independently of tetramer formation and septal anchoring in Vibrio cholerae
The study of Vibrio cholerae MatP reveals the degree of integration of the plasmid-derived chromosome carried by the bacterium, and suggests that the ancestral role of MatP is to promote cell division at the end of the chromosome replication and segregation cycle. V. cholerae harbours a primary chromosome derived from the monochromosomal ancestor of the Vibrionales (ChrI) and a secondary chromosome derived from a megaplasmid (ChrII). The coordinated segregation of the replication terminus of both chromosomes (TerI and TerII) determines when and where cell division occurs. ChrI encodes a homolog of Escherichia coli MatP, a protein that binds to a DNA motif (matS) that is overrepresented in replication termini. Here, we use a combination of deep sequencing and fluorescence microscopy techniques to show that V. cholerae MatP structures TerI and TerII into macrodomains, targets them to mid-cell during replication, and delays their segregation, thus supporting that ChrII behaves as a bona fide chromosome. We further show that the extent of the segregation delay mediated by MatP depends on the number and local density of matS sites, and is independent of its assembly into tetramers and any interaction with the divisome, in contrast to what has been previously observed in E. coli. More information: https://www.nature.com/articles/s41467-024-54195-0 Contact: E. Galli elisa.galli@i2bc.paris-saclay.fr & FX Barre francois-xavier.barre@i2bc.paris-saclay.fr
Le prochain café se tiendra le vendredi 22 novembre 2024 en visioconférence de 13h30 à 14h avec la participation de Denis Faure (I2BC) qui nous parlera sur « The versatile lifestyle of the plant pathogen Agrobacterium tumefaciens » Pour assister en visioconférence : ICI
Via Life Sciences UPSaclay
|







![Complete description of the biosynthetic process of [2Fe-2S] clusters | I2BC Paris-Saclay | Scoop.it](https://img.scoop.it/THKpAN1-eQiW-1DEJF_ZmDl72eJkfbmt4t8yenImKBVvK0kTmF0xjctABnaLJIm9)