Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:35 PM
|
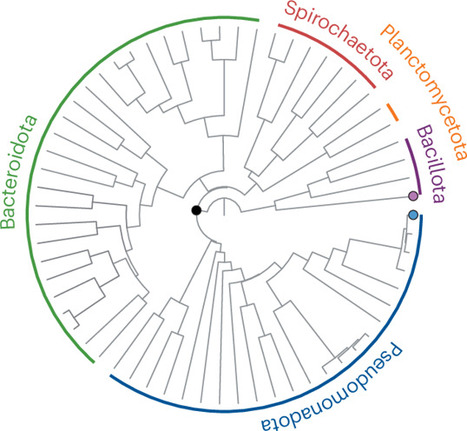
Predicting how metabolically interdependent microbial communities will respond to stress is an important but difficult challenge. Synthetic microbial communities provide an important tool for identifying mechanisms that impact stress response in communities. In this review, we discuss the weakest-link hypothesis as a null model that predicts that all species in an interdependent community will be constrained by the species that is most sensitive to a given stress. The hypothesis is contingent on the assumptions that 1) stress does not alter metabolite exchange in a way that changes growth constraints, 2) cross-feeding does not alter the ability of cells to withstand stress, and 3) the amount of stress experienced by a species is not altered by partners through mechanisms unrelated to cross-feeding. We then present cases that violate these assumptions to highlight the complexity of ecological factors involved in community stress response. Finally, we discuss the evolutionary dynamics of cross-feeding microbial systems when exposed to stress and some important challenges for connecting work from synthetic communities to more complex systems.
|
|
Scooped by
mhryu@live.com
Today, 2:26 PM
|
The biosynthesis of surfactin, a potent lipopeptide biosurfactant produced by Bacillus subtilis, imposes a significant metabolic burden on the host organism. Understanding the metabolic costs associated with surfactin production, specifically the ATP demand and precursor diversion resulting from the expression of the large srfAA-AD operon (srfA operon), is crucial for optimizing production strains. In this study, the metabolic burden and growth impacts associated with surfactin biosynthesis in B. subtilis were quantitatively evaluated by comparing a reference surfactin-producing strain (BMV9) with two mutant strains: BMV12, which lacks the srfA operon, and BMV33, which retains the srfA operon but lacks the sfp gene. Our analysis included theoretical calculations of ATP and NADPH + H+ requirements for de novo surfactin synthesis, and we measured growth behavior and carbon and nitrogen source consumption. Results indicated that surfactin production significantly reduces biomass yield (YX/S) and specific growth rates due to the metabolic costs of expressing the non-ribosomal peptide synthetase (NRPS) and the diversion of key precursors. Notably, BMV12 exhibited higher growth rates compared to the surfactin-producing strain. Proteome analyses further revealed differential protein abundance in non-surfactin-producing strains, indicating altered metabolic pathways that may relieve the metabolic burden associated with surfactin synthesis. These findings highlight the complex trade-offs between secondary metabolite production and cellular growth, providing a foundation for metabolic engineering strategies aimed at optimizing surfactin yields while minimizing metabolic costs.
|
|
Scooped by
mhryu@live.com
Today, 2:21 PM
|
Ammonia oxidation is a rate-limiting step in the nitrogen cycle, yet viral contributions to this process remain largely unresolved. Here, we identify three genomically distinct groups of amoC-encoding phages (155-338 kilobases in length; termed as amoC-phages) from multiple freshwater lakes in Europe and North America, including the Laurentian Great Lakes. These phages are highly divergent in phylogeny, genome architecture, and gene content, and are predicted to infect two distinct Nitrosomonadaceae ammonia-oxidizing bacterial lineages. The placement of phage-encoded amoC genes across these divergent viral clades indicates independent acquisition of amoC. Time-series and depth-resolved metagenomes and metatranscriptomes reveal persistent and depth-structured distributions of amoC-phages and their predicted hosts, with seasonal mixing periodically reshaping their co-occurrence patterns. Furthermore, virome data from Lake Mendota show that some of the amoC-phages occur as free viral particles, supporting active viral lysis and particle redistribution along the water column. Metatranscriptomes of the Laurentian Great Lakes reveal coordinated expression of phage structural genes (e.g., major capsid protein) together with phage-encoded amoC, indicating active infection in situ. Together, these results support a framework in which amoC-phage infection is depth-structured, seasonally dynamic, and coupled to ammonia-oxidizing bacterial host activity, highlighting viruses as previously overlooked components of freshwater nitrogen cycling.
|
|
Scooped by
mhryu@live.com
Today, 2:05 PM
|
Wine fermentation remains inherently variable because of the genetic and phenotypic diversity of Saccharomyces cerevisiae and non-Saccharomyces yeasts, microbial interactions, and climate-driven shifts in grape composition, challenging predictable and low-intervention winemaking. Recent population genomics advances, including telomere-to-telomere assemblies and pan-genome analyses of yeast genomes, have transformed the field by revealing structural variation, introgressions, hybridization, and gene content diversity underlying key enological traits. High-throughput functional genomics, quantitative trait locus mapping, multi-omics, and machine learning increasingly connect these features with fermentation kinetics, stress tolerance, and aroma biosynthesis. The near completion of the Synthetic Yeast Genome (Sc2.0) and its SCRaMbLE system further expands the experimental design space for rapid genome rearrangement and strain innovation. These advances have improved identification of candidate determinants of industrially relevant phenotypes, but robust genotype-to-phenotype prediction remains limited by polygenic architectures, epistasis, environmental dependence, and microbial context. Future progress will depend on integrating population genomics with functional validation, realistic phenotyping, and interpretable predictive frameworks to support rational yeast engineering and more consistent, sustainable winemaking.
|
|
Scooped by
mhryu@live.com
Today, 1:59 PM
|
Serine integrases enable precise genome engineering but remain underused for iterative genome integration because current workflows are often labor-intensive and difficult to scale. Here, we report SARGE, a serine integrase-assisted rapid genome integration platform, that enables iterative insertion of large DNA fragments into the E. coli genome through cassette exchange. By combining the orthogonal serine integrases PhiC31 and Bxb1 with rational donor plasmid design, SARGE supports rapid, programmable integration cycles without the need for resistance marker excision between rounds. To simplify the identification of correct recombinants, we incorporated a dual-fluorescence reporter system based on sfGFP and mKate and developed a green–red screening strategy for direct, naked-eye colony selection. This visual screen identified the desired recombinants with 100% accuracy among the colonies tested. SARGE achieved cassette exchange efficiencies of up to 95% and maintained efficiencies of approximately 90% for cargoes as large as 10 kb. Together, these features substantially streamline iterative genome integration and establish SARGE as a robust and accessible platform for genome engineering and synthetic biology in E. coli, with potential for extension to other genetically tractable microbial hosts.
|
|
Scooped by
mhryu@live.com
Today, 1:50 PM
|
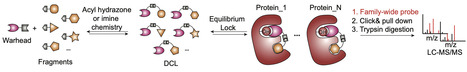
Mass spectrometry-based chemical proteomics enables unbiased assessment of ligand potency and selectivity across the proteome. However, current approaches remain limited by the low throughput of single-compound screening and reliance on pre-synthesized libraries. Here we devise a mechanism-driven “library-versus-proteome” platform that couples dynamic combinatorial libraries with activity-based protein profiling, enabling real-time selection and optimization of ligands in complex biological systems. This approach increases screening throughput by 10- to 20-fold, streamlines library generation, and adopts a “screen first, synthesize later” paradigm. Applying this platform, we discover covalent inhibitors of serine hydrolases including PPME1, ABHD11 and PNPLA6, and reveal uncharacterized roles of PNPLA6 in lipid metabolism and cancer cell proliferation. We further extend the strategy to cysteine-targeting ligands by designing tailored warheads, enabling proteome-wide EC50 profiling of over 2600 ligandable cysteines and yielding inhibitors for NIT2, PRDX5, TXNDC17 and VCP. Focusing on VCP, we uncover a previously unrecognized signaling axis in which GPCR activity modulates activation of the ER stress-induced unfolded protein response. Using a gel-based “library-versus-proteome” assay, we screen over 800 analogues within two days and identify a more potent VCP ligand with nanomolar activity and in vivo antitumor efficacy. This work establishes library-versus-proteome screening as a scalable strategy for ligand discovery. Chemical proteomics can reveal how small molecules act across the proteome, but current single-compound screens are slow and require premade libraries. Here, the authors develop a library-versus-proteome platform that rapidly discovers covalent ligands and potent inhibitors of cancer-relevant targets.
|
|
Scooped by
mhryu@live.com
Today, 1:26 PM
|
Lysine is an essential amino acid often limited in plant-based foods, making its enrichment in fermented foods a key nutritional goal. Bacillus and Priestia species are natural lysine producers, but their biosynthetic capacity is constrained by riboswitch regulation of the aspartokinase gene lysC. In this study, we employed mutagenesis using the lysine analogue aminoethylcysteine (AEC) to generate lysine-overproducing strains. AEC-resistant mutants carried distinct mutations in the lysC riboswitch and in lysine biosynthetic and transporter genes. Several mutants showed significantly increased lysine production in defined media. One riboswitch mutant produced over 150-fold more free lysine than its wild type in defined media and threefold during oat fermentation, establishing proof of concept for lysine enrichment in food matrices. Structural predictions revealed that lysC riboswitch mutations occurred near the lysine binding pocket, suggesting altered ligand-binding dynamics. These findings highlight riboswitch-targeted mutagenesis as a promising non-recombinant strategy to enhance lysine production in food fermentations.
|
|
Scooped by
mhryu@live.com
Today, 12:44 PM
|
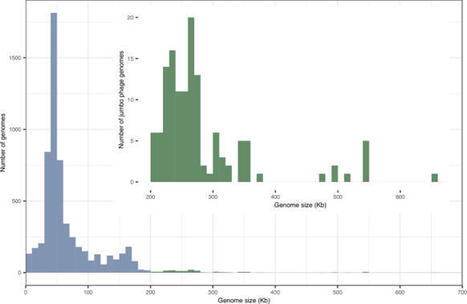
Jumbo bacteriophages are bacterial viruses with double-stranded DNA genomes exceeding 200 kb. These viral giants feature exceptionally large virions, expansive genetic repertoires, and in some cases, remarkable eukaryotic-like traits. Jumbo phages challenge long-standing notions of phage simplicity, redefining the boundaries of what a phage can be. In this Review, we examine the biology of jumbo bacteriophages, highlighting their diversity, evolutionary origins, distinctive morphologies and lifecycles, complex interactions with bacterial hosts, and their potential for biotechnology and therapy, with a focus on, but not limited to, the Chimalliviridae phages. Jumbo phages are bacterial viruses with double-stranded DNA genomes exceeding 200 kb. In this Review, Sam van Beljouw & Li Yuping examine the biology of these bacteriophages, highlighting their diversity, evolutionary origins, distinctive morphologies and lifecycles, interactions with bacterial hosts, and their potential for biotechnology and therapy.
|
|
Scooped by
mhryu@live.com
June 19, 5:07 PM
|
RNAs adopt complex structures that regulate key biological processes, making accurate structure prediction essential. Chemical probing coupled with Nanopore direct RNA sequencing (DRS) offers a route to single-molecule structural inference, but current tools are limited by inaccurate signal-to-sequence alignment, which degrades modification-rate estimation and downstream structure prediction. Here we introduce segSHAPE for RNA secondary structure prediction from Nanopore DRS data (both RNA002 and RNA004 chemistries), a probe-agnostic framework that improves signal alignment using prior information of basecalling and per-read signal baseline shift correction, learns position-specific k-mer raw signal parameters, and estimates per-nucleotide modification rates with an unsupervised anomaly detector. On three public RNA002 DRS datasets spanning different chemical probes (AcIm, NAI-N3) and RNAs from 421 to 1552 nt, segSHAPE achieves the highest F1 score and Matthews correlation coefficient (MCC) on all RNAs, exceeding the strongest baseline by 3.4 to 5.8 percentage points in MCC. It additionally captures the ligand-induced conformational change of the thiamine pyrophosphate (TPP) riboswitch RNA directly from RNA002 DRS data using the DEPC probe. On a public RNA004 DRS dataset, segSHAPE improves over the sm-PORE-cupine baseline by 17 ROC-AUC points in modification rate estimation and by 6.7 MCC points in structure prediction. These results establish segSHAPE as a unified, probe-agnostic pipeline for RNA structure prediction from Nanopore DRS data.
|
|
Scooped by
mhryu@live.com
June 19, 3:52 PM
|
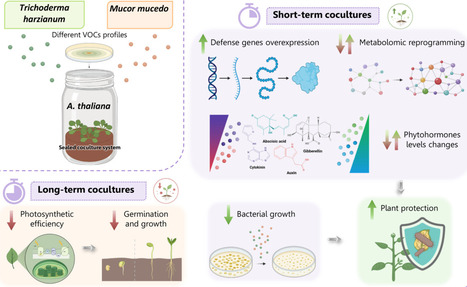
Volatile organic compounds (VOCs) emitted by certain fungi have previously been suggested to modulate plant development and their response to stresses. While some fungal species, such as Trichoderma harzianum, are known to produce VOCs, the production of VOCs in other phylogenetically divergent species, such as Mucor mucedo, was previously unknown. This study aimed to analyze the VOCs profiles emitted by these fungal species and examine their effects on the growth and defensive responses of Arabidopsis thaliana. Volatilome analysis revealed that the most abundant VOCs for both species were alcohols, ketones and esters. However, notable differences were also observed, particularly with regard to terpenes. We then observed that prolonged exposure to VOCs emitted by both fungal species had detrimental effects on A. thaliana growth and photosynthetic performance, whereas shorter exposures enhanced the expression of defense-related genes and the plant defense against the phytopathogenic bacterium Pseudomonas syringae. This increased resistance does not appear to be mediated by canonical H2O2-induced immunity, but rather by subsequent responses that trigger complex metabolic reprogramming, including glucosinolate biosynthesis. Therefore, our results confirm that VOC-mediated plant-fungus interactions are very relevant to plant fitness, highlighting the need to understand them in greater depth.
|
|
Scooped by
mhryu@live.com
June 19, 3:45 PM
|
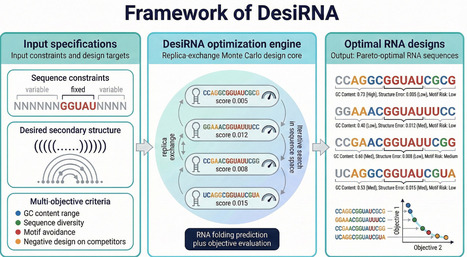
Programs and computational frameworks for predicting RNA sequences with desired folding properties are continually being developed and expanded. A decade has passed since they were last reviewed in this journal, and this brief review provides an update to the review published at that time. Given a target secondary structure, these programs aim to predict RNA sequences that fold into the desired structure while satisfying various constraints. This procedure is known as inverse RNA folding. Traditionally, inverse RNA folding has been used to design optimized RNAs with favorable properties. This updated review covers some of the most widely used freeware programs developed for this purpose over the past decade. RNAinverse, part of the Vienna RNA package, was the first program devised to address the inverse RNA folding problem, and many subsequent programs were described in the earlier review. Some of the most important computational frameworks are the Infrared framework and DesiRNA. In addition, RNA design capabilities have been incorporated into the RNAstructure package, while NUPACK, as well as MoiRNAiFold, MODENA, incaRNAfbinv, and related tools have undergone recent updates. A variety of strategies have also emerged to address the problem of 3D RNA design and RNA–RNA interactions. The various programs mentioned employ distinct approaches, ranging from replica exchange Monte Carlo to constraint satisfaction, as well as Boltzmann sampling and machine learning approaches. Machine learning methods are being developed for emerging applications in biotechnology such as messenger RNA(mRNA) design and CRISPR guide RNA (gRNA) design. This brief review examines these programs and provides a timely update.
|
|
Scooped by
mhryu@live.com
June 19, 3:29 PM
|
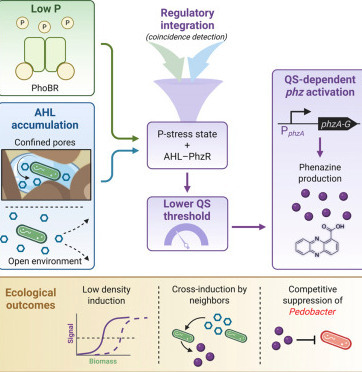
Bacteria often coordinate collective behaviors such as biofilm formation and secondary metabolite production through quorum sensing (QS), a regulatory system traditionally linked to high cell density. However, in environments such as soil, where microbial populations are spatially fragmented, sparse, nutrient-limited, and subject to mass transport, the mechanisms that enable QS-dependent processes remain incompletely understood. Here, we investigate the regulation of a secreted redox-active metabolite, phenazine-1-carboxylic acid (PCA), in Pseudomonas synxantha 2-79, a model rhizobacterium, under phosphorus (P) limitation, a persistent stress in many soils. Using a combination of microscopy and molecular genetic approaches, we show that P limitation sensitizes the QS activation threshold by an order of magnitude, enabling phenazine induction at relatively low population densities compared with P-replete conditions. This induction is abolished in QS-deficient mutants and restored by the addition of exogenous acyl-homoserine lactone (AHL), demonstrating that QS remains essential, but its threshold becomes environmentally tuned. Under P limitation, spatial confinement and pore saturation levels further shape the timing and location of induction, illustrating how physical structure and nutrient stress can modulate bacterial activities. Moreover, P stress confers both collaborative and competitive advantages, enabling P. synxantha to undergo low-cell-density AHL cross-induction with related Pseudomonas spp. and to suppress other rhizobacteria. Lastly, on plant roots, phenazine biosynthetic genes are more strongly induced under P limitation. These findings illustrate how the nutrient status of an environment can modulate the onset of QS, enabling quorum-regulated behaviors to activate at lower thresholds.
|
|
Scooped by
mhryu@live.com
June 19, 3:02 PM
|
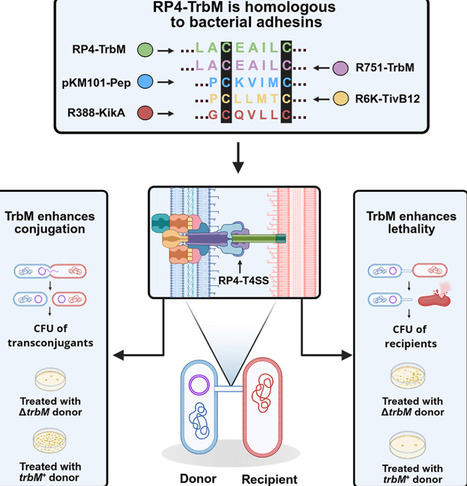
Bacterial conjugation drives horizontal gene transfer and antibiotic resistance via type IV secretion systems (T4SS) on conjugative plasmids like RP4. Previously, we found that the RP4-T4SS mediates interbacterial killing, a process enhanced by the uncharacterized gene trbM. Here, we characterize RP4-TrbM and identify structural features essential for boosting both conjugation and killing. Computational analyses reveal that the RP4-TrbM shares similarities with known bacterial adhesins in other conjugative systems. Homologs from plasmids R751, R388, and pKM101 could complement RP4-TrbM-knockout strains, restoring and enhancing conjugation (R751-TrbM and pKM101-Pep) and conjugation-associated killing (R751-TrbM, R388-KikA, and pKM101-Pep), while TivB12 from conjugative plasmid R6K could not complement the RP4-T4SS. Furthermore, we identified an essential functional domain in RP4-TrbM that retains activity even when repositioned between a different signal peptide and C terminus. These findings expand our understanding of the RP4 conjugative machinery and highlight TrbM-like proteins as promising targets for inhibiting T4SS-mediated processes.
|
|
|
Scooped by
mhryu@live.com
Today, 2:28 PM
|
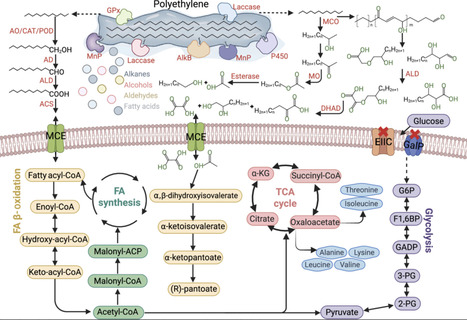
The growing global plastic waste crisis demands the development of urgent, effective, and sustainable solutions. While conventional recycling methods present intrinsic limitations, microbial biodegradation of plastic waste has emerged as a promising alternative. In this review, we explore the potential of using microorganisms to degrade major hydrocarbon-based plastic polymers and discuss key aspects of this rapidly advancing field, including (i) isolation and characterization of novel microorganisms and enzymes in hydrocarbon-based plastic biodegradation, (ii) development and streamlining of microbial consortia to improve hydrocarbon-based plastic biodegradation efficiency, and (iii) investigation of natural biodegradation processes to illustrate the relationship between plastic degradation and environmental influence. We highlight practical biotechnological approaches and advanced computational tools in hydrocarbon-based plastic degradation, as hydrocarbon-based plastic represents the highest proportion of plastic waste while still lacking effective conversion strategies. Our ultimate goal is to integrate microbial biodegradation strategies into modern waste-management systems and offer a feasible pathway toward a circular bioeconomy, one in which persistent plastic polymers are no longer treated as waste but are converted into renewable feedstocks that support sustainable resource recovery.
|
|
Scooped by
mhryu@live.com
Today, 2:24 PM
|
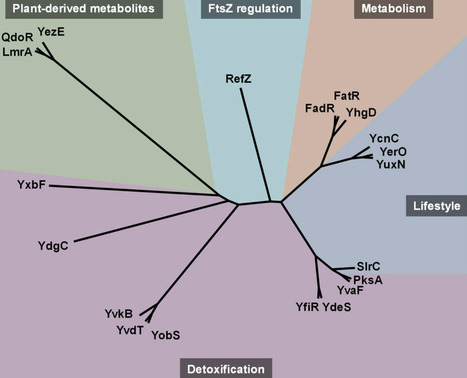
Although the Gram-positive bacterium Bacillus subtilis is one of the best studied model organisms, there remain many genes and proteins whose functions are unknown. This is also true for many transcriptional regulators that are key to adaptation to changes in the environment. One class of regulators is the group of TetR family of regulators (TFRs). The genome of B. subtilis encodes 20 TFRs, while more than half are poorly characterized. Characterized TFRs of B. subtilis participate in many different aspects of bacterial physiology, ranging from regulation of metabolism, response to plant-derived compounds, detoxification, lifestyle changes, and regulation of septum formation during sporulation. Here, we summarize what is known about the studied regulators and how we can use the knowledge of the characterized TFRs in concert with novel approaches to investigate the function of the poorly characterized regulators. In many cases, gene synteny, meaning the co-conservation of two or more genes together across different species, is a helpful tool to assess functional relationships. Furthermore, structural homologies of the TFRs can hint at similar repressor-ligand interactions and thus also contribute to formulating hypotheses about processes in which they might be involved. Moreover, with the help of innovative tools that predict protein-protein or protein-DNA interactions, we can develop hypotheses about molecular regulatory mechanisms and design experiments based on them. Finally, we discuss some of the more unique TFRs of B. subtilis and propose experiments to elucidate their regulatory functions and the processes they participate in.
|
|
Scooped by
mhryu@live.com
Today, 2:12 PM
|
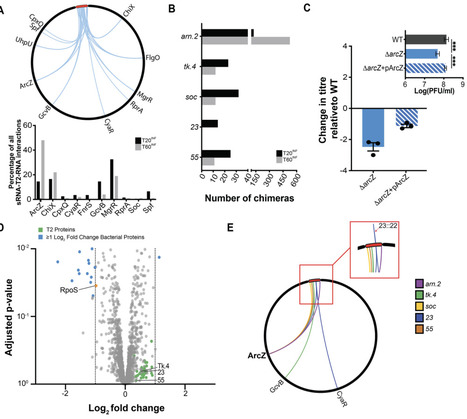
Host acquisition by bacteriophages often entails modulation, appropriation, or inhibition of components and processes central to bacterial gene expression. Small non coding RNAs (sRNAs) are major regulators of RNA fate and frequently rely on the conserved RNA chaperone Hfq to engage their cognate targets. Although phages are known to encode specialised proteins and sRNAs to manipulate host gene expression, it has remained unclear whether they also co opt host encoded sRNAs for their own regulatory needs. We show that transcriptome wide Hfq mediated RNA-RNA interactions are broadly destabilised during T2 phage infection of E. coli. We further demonstrate that the conserved bacterial sRNA ArcZ is co-opted by T2 to promote expression of a conserved phage operon that includes a protein which inhibits a bacterial restriction-modification system. ArcZ achieves this by preventing RNase E-mediated degradation of the transcript originating from the phage operon. Our study provides the first evidence of an evolutionary strategy in which a phage leverages a nucleic acid host factor to fulfil its own gene expression requirements.
|
|
Scooped by
mhryu@live.com
Today, 2:03 PM
|
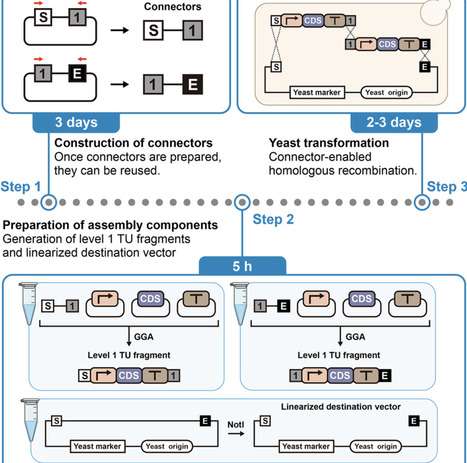
Here, we present a protocol for multigene pathway construction in Saccharomyces cerevisiae using a connector-enabled workflow that integrates Golden Gate Assembly with yeast homologous recombination. We provide procedures for assembling Level 1 transcriptional unit (TU) fragments using standardized connectors and directly co-transforming them into S. cerevisiae to form Level 2 multigene plasmids. Compared to conventional approaches, this protocol significantly reduces procedural complexity, minimizes dependence on operator expertise, and substantially accelerates workflow timelines.
|
|
Scooped by
mhryu@live.com
Today, 1:56 PM
|
Natural product synthesis is key to unravel the roles and functions of complex biological molecules and drive innovations in drug discovery, agrochemicals, and materials sciences. Flavonoids are ubiquitous plant secondary metabolites with a C(6)–C(3)–C(6) benzo-γ-pyrone carbon skeleton, and they encompass a vast family of derivatives that play essential roles in UV protection, flower pigmentation, auxin transport, and defense against environmental stress. Flavonoid biosynthesis yields a diverse array of compounds, including flavones, anthocyanins, and proanthocyanidins, whose stability and bioactivity are often enhanced by post-translational modifications such as glycosylation and methylation. Flavonoids are known for their potent antioxidant properties and thus play a critical role in neutralizing reactive oxygen species and preserving cellular redox balance. Beyond their antioxidant activity, these phytochemicals exhibit a wide range of biological effects, including antibacterial, antiviral, anti inflammatory, and anticancer activities, highlighting their significant therapeutic potential. Due to their structural complexity and pharmacological promise, the total synthesis of flavonoids and their glycosylated analogues has garnered considerable research interest. This review aims to provide an overview of the recent advances in the total synthesis of flavonoid glycosides and their derivatives over the last decade. We selected twenty exemplary examples to illustrate key synthetic strategies while discussing their natural sources, therapeutic applications, and structure–activity relationship (SAR) studies that helped elucidate specific functional groups that are important for their pharmacological properties. We hope we can provide a current perspective on the recent advancements in flavonoid chemistry and their significance in the development of novel therapeutic agents for a range of diseases.
|
|
Scooped by
mhryu@live.com
Today, 1:31 PM
|
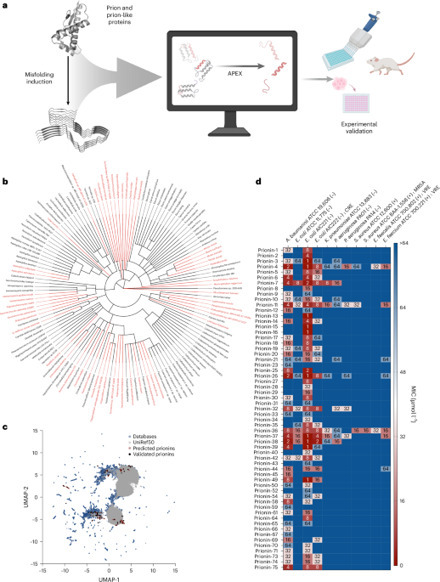
Prion and prion-like proteins are classically associated with protein misfolding, but amyloidogenic sequences can also participate in host defence. Here, using deep learning, we screened 19.3 million fragments from 2,897 curated prion-related proteins and identified 1,179 candidate antimicrobial peptides, which we term prionins. Among 75 synthesized prionins, 59 inhibited bacterial pathogens, 53 perturbed membranes and 2 reduced Acinetobacter baumannii infection burden in mice. Deep learning reveals that prion-related proteins contain hidden ‘prionin’ peptides with antibacterial activity, including two candidates that reduced infections in mice.
|
|
Scooped by
mhryu@live.com
Today, 1:19 PM
|
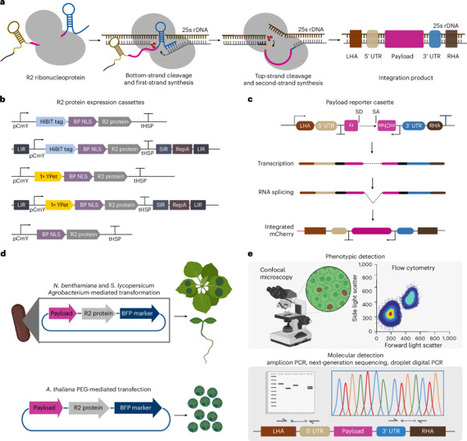
Traditional approaches for DNA insertion into plant genomes using Agrobacterium tumefaciens result in random integration. Newer genetic engineering methods based on nucleases, prime editors, transposases and recombinases extend capabilities but remain constrained with low efficiencies, off-target integration or limited payload size. Here we adapt the avian Taeniopygia guttata R2 protein (R2Tg) for targeted DNA insertion into plant genomes by engineering R2Tg expression cassettes and RNA payloads carrying intron-disrupted reporters, with optimized ribosomal DNA homology arms and untranslated regions. In Arabidopsis thaliana protoplasts, Nicotiana benthamiana leaves and Solanum lycopersicum seedlings, our R2Tg editor system achieves targeted insertion of full-length payloads ranging from 2.2 kb to 5 kb. In Nicotiana benthamiana leaves, integration occurs, on average, at 1 copy per genome, which is 30 times more efficient than that achieved by Cas9 homology-directed repair. This work establishes an R2Tg ribonucleoprotein platform for targeted DNA insertion into plant genomes, using a multicopy genomic safe-harbor site to enable efficient addition of multikilobase genes. R2 retrotransposons are used to integrate DNA into plant and crop 25S ribosomal DNA sites.
|
|
Scooped by
mhryu@live.com
June 19, 5:21 PM
|
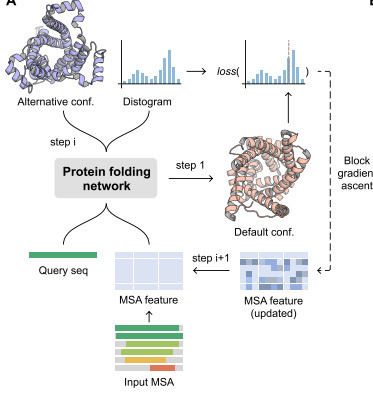
End-to-end structure predictors, such as AlphaFold2, typically output only the dominant conformational state of a given protein, which is biased by the training data set. Existing strategies for recovering alternative conformations are often computationally expensive and offer limited biological interpretability. Here, we present SteerAF, an inference-time optimization framework based on AlphaFold2 that leverages information encoded in the distogram derived from deep multiple sequence alignments (MSAs) to predict alternative protein conformations. Across four benchmark datasets, SteerAF matches or surpasses existing methods in predicting alternative conformations for the majority of systems. Sparse MSA-feature modifications generated via block gradient ascent exhibit a strong correlation with experimentally characterized functional residues, recovering them with approximately 50% precision in the tested proteins. Furthermore, SteerAF enables effective decoy selection in the absence of experimental structures, and its predictions can serve as seed structures for molecular dynamics simulations to map conformational landscapes. Thus, SteerAF provides an efficient and interpretable approach for predicting alternative conformations, offering a framework that can be extended to other similar predictors and problems.
|
|
Scooped by
mhryu@live.com
June 19, 4:37 PM
|
Phenotypic biosensors that measure bacterial viability and antimicrobial susceptibility are essential for rapid infectious disease diagnostics, yet their speed is fundamentally limited by the rate at which bacteria encounter reporter molecules, a transport bottleneck that has been typically addressed by complex microfluidic solutions. Here we show that this bottleneck can be overcome by engineering transport directly into the sensing material. A multifunctional ionic hydrogel matrix, co-encapsulating bacterial growth medium and the redox reporter resazurin, exploits swelling-driven convective transport to dramatically accelerate bacteria-reporter interactions without any change to assay chemistry. By systematically tailoring the hydrogel crosslinking density and optimizing the encapsulated nutrient-osmotic microenvironment, we maximize metabolic signal generation to achieve a 12-to-48-fold reduction in detection time relative to solution-phase and conventional hydrogel assays. Deployed in a standard 96-well format for urinary tract infection (UTI) diagnosis of 48 clinical samples, the platform rapidly detects infection in 15 minutes to 2 hours, achieving 95% sensitivity and 100% specificity for bacterial detection, and 100% sensitivity and 98% specificity for antimicrobial susceptibility profiling, compared to time-consuming gold-standard urine culture-based methods. Results are readable both quantitatively on a plate reader and visually as a colorimetric assay, enabling point-of-care deployment without additional instrumentation. Thus, embedding transport enhancement within the sensing matrix, represents a general and scalable design principle for accelerating interaction-limited biosensing, which has excellent scope for rapid diagnostic development.
|
|
Scooped by
mhryu@live.com
June 19, 3:49 PM
|
Methanol oxidation by alcohol oxidases (AOXs) is a key bottleneck in one-carbon (C1) bioconversion due to limited catalytic efficiency and poor formaldehyde tolerance. Here, we report the directed evolution of an alcohol oxidase from Gloeophyllum trabeum, yielding an optimized variant, GtAOXM3. The engineered enzyme exhibits a sixfold increase in catalytic efficiency (7.7 s−1 mM−1), together with enhanced formaldehyde tolerance, thermostability, and high methanol specificity. Molecular dynamics simulations suggest that increased global rigidity and cooperative residue dynamics contribute to the improved performance. When incorporated in multienzyme cascade systems, GtAOXM3 enables efficient conversion of methanol to the value-added chemicals dihydroxyacetone (34.5 mM) and ethylene glycol (23.3 mM). This work establishes GtAOXM3 as an efficient and cost-effective biocatalyst for methanol-based C1 biotransformation.
|
|
Scooped by
mhryu@live.com
June 19, 3:35 PM
|
The immunosuppressive tumor microenvironment (TME), driven by lactate accumulation, critically limits cancer immunotherapy efficacy. In this research article, we engineered Shewanella oneidensis MR-1 by reprogramming energy metabolism to reverse lactate-driven immunosuppression in the TME. Integration of polyphosphate kinase 2 and NAD kinase markedly augmented bacterial bioenergetics, boosting ATP production by 287.5% and elevating the NADH/NAD+ ratio by 299.9% compared with the ‘wild-type’ (WT) strain. This bioenergetic enhancement accelerated targeted lactate depletion and increased intratumoral bacterial persistence, while impairing tumor lactate transport via downregulation of monocarboxylate transporters MCT1 and MCT4. Crucially, metabolic remodeling reversed immunosuppression by enhancing antigen presentation and CD8+ T cell infiltration, and reducing regulatory T cells, thereby converting immunologically ‘cold’ tumors into immunoreactive phenotypes. When combined with anti-programmed cell death protein 1 (αPD-1) immunotherapy, this strategy synergistically amplified antitumor efficacy and established durable immunological memory against recurrence. Collectively, our work establishes a paradigm for bacterial bioenergetic engineering to advance cancer immunotherapy.
|
|
Scooped by
mhryu@live.com
June 19, 3:14 PM
|
Phages, the most abundant biological entities on Earth, infect bacteria and reprogram them into ‘virocells’ with altered physiology and ecology. While metagenomic studies have largely inferred reprogramming through virus-encoded auxiliary metabolic genes (AMGs), phages can reprogram cells through many other tools. In this review, we explore how phage-encoded, host-acting transcription and sigma factors (TSFs) reshape host transcriptional networks beyond simply regulating phage replication. We synthesize emerging genomic evidence for TSF prevalence in phages, mechanistic insights into how host-acting TSFs might influence ecologically relevant cellular functions, and highlight recent experimental and bioinformatic advances that make TSFs particularly tractable for large-scale bioinformatic studies. Together, we position TSFs as AMG-complementary mechanisms of viral reprogramming, tractable for metagenomic inferences, with potential cellular- and ecosystem-level consequences that can power translational applications.
|