 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:19 PM
|
Optogenetic methods are powerful tools for synthetic biology, allowing light to control cellular processes. While most bacterial optogenetic systems regulate gene expression at the transcriptional level, relatively few enable post-translational control, which can provide faster and growth-independent regulation of protein activity. Here, we describe the development of a post-translational optogenetic tool in E. coli using the Mesoplasma florum Lon (mfLon) protease, an AAA+ protease that is orthogonal to native E. coli degradation machinery. To engineer a light-responsive mfLon, we constructed a large library in which a blue-light responsive domain, Avena sativa LOV2, was introduced into nearly every codon position in the protease using an unbiased molecular approach. We screened 726 mfLon-LOV variants using fluorescence-activated cell sorting and multi-round enrichment campaigns. We identified a novel dark-active variant (mfLon-LOV-534) that degrades target proteins in the dark and is inactivated upon blue-light exposure. Characterization of this variant demonstrates that its proteolytic response can be tuned by varying blue-light intensity and transcriptional expression levels. Furthermore, we show that mfLon-LOV-534 can degrade a target protein in both exponential and stationary growth phases, which addresses the limitations of division-based protein dilution. This work establishes a scalable approach to engineering allosteric control in complex multimeric enzymes and provides a foundation for orthogonal, growth-independent control of protein stability in synthetic circuits.
|
|
Scooped by
mhryu@live.com
Today, 4:23 PM
|
Bacterial colonies grow within microenvironments that they continuously reshape through nutrient uptake, metabolism and mechanical interaction. However, in colonies carrying engineered gene circuits, how these self-generated environmental changes feed back on gene expression to produce spatiotemporal organization remains poorly understood. Here we show that growth and gene expression are dynamically coupled during the maturation of founding colonies, with growth-driven environmental changes organizing gene expression into traveling waves. By combining quantitative time-lapse microscopy, mathematical modeling and image-based parameter inference, we demonstrate that edge-dominated colony expansion consistent with mechanical constraints on growth is followed by density-dependent growth arrest in which the total area occupied by colonies does not converge to a fixed carrying capacity of the shared growth environment. We then find that colonies form two distinct traveling waves of gene expression. An intra-colony wave emerges only after growth arrest and is observed in both constitutive genes and distinct regulated circuit architectures, indicating that it is not a result of circuit topology. Our observations are consistent with nutrient depletion during growth followed by subsequent recovery after growth arrest. At later times, an inter-colony wave emerges that is consistent with a colony-produced diffusible factor spreading through the shared medium. Together, these findings reveal that the colony environment is not a passive background but an active and intrinsic component of spatiotemporal gene regulatory dynamics, in which self-generated environmental feedback couples mechanical constraints, nutrient dynamics and gene expression across spatial scales.
|
|
Scooped by
mhryu@live.com
Today, 4:02 PM
|
Precise regulation of protein synthesis is fundamental to cellular homeostasis and remains a primary target for synthetic biology applications. However, the non-linear relationship between mRNA abundance and protein levels presents complexities that poses challenges for predictive engineering. Here, we present TRIM, a Transformer-based RNA Inference Model that leverages full-length mRNA sequences and multi-omics data to predict translation efficiency. By employing a Parallel Expert Mixer, TRIM achieves robust prediction accuracy (R2 ≥ 0.8,Pearson r ≥ 0.9). Trained on multimodal data from massive Saccharomyces cerevisiae isolates, TRIM demonstrates outstanding biological interpretability, helping to decipher complex translational patterns such as synergistic effects between bases, sequence-dependent codon preference in different stages, and distinct attention on key secondary structures. These results indicate that the integration of multi-omics data with holistic sequence modeling can effectively decode the cis-regulatory grammar of translation as well as providing a scalable and interpretable generative framework for future synthetic biology engineering.
|
|
Scooped by
mhryu@live.com
Today, 3:42 PM
|
The mutational spectrum is an increasingly important molecular phenotype that quantitatively describes mutagenesis in a given gene and species, enabling future comparative analyses to reveal differences in underlying mutagenic processes, whether internal, such as DNA repair processes, or external, such as ecological niches and conditions. Mutation accumulation experiments, although time-consuming and costly, remain the standard approach for reconstructing bacterial neutral mutation spectra. Here, we present BacNeMu, a phylogenetically informed pipeline that reconstructs neutral mutational spectra of bacterial genomes using open databases GTDB, AnnoTree and KEGG Orthology, building on previously developed NeMu pipeline. BacNeMu reconstructs mutation spectra that closely match mutation accumulation experiments results while requiring substantially less time, enabling comparative analyses across diverse bacterial taxa. Applied to obligate aerobes and anaerobes, BacNeMu recovered the expected excess of T:A>C:G transitions, consistent with oxidative-damage-associated mutational patterns previously described in mitochondrial genomes and yeast single-strand. We further asked if any other ecologic factors influence a mutational spectrum. As a pilot we compared three species living under different temperatures: one strong thermophile - Thermotoga maritima, one psychrophile - Clostridium algidicarnis, and one with intermediate temperature tolerance - Psychrobacter sanguinis. In the thermophile, the relative frequency of T:A>C:G substitutions was higher than in the psychrophile, consistent with the hypothesis that GC-biased mutagenesis contributes to thermal adaptation, although C:G>T:A transitions predominate across all three species. BacNeMu provides a rapid, phylogenetically informed framework for generating biologically meaningful mutation spectra from open databases.
|
|
Scooped by
mhryu@live.com
Today, 2:47 PM
|
Neisseria gonorrhoeae is a common Gram-negative pathogen with increasing resistance to all recommended antibiotics. There is a critical need to improve the efficiency of the antibiotic hit discovery process to replenish the drug development pipeline. Here, we show that deep learning models can augment high-throughput screens to identify readily available molecules with narrow-spectrum activity against difficult-to-treat strains of N. gonorrhoeae. We phenotypically tested 38,650 small molecules for N. gonorrhoeae growth inhibition to train a predictive graph neural network (GNN) model. We benchmarked the model’s performance against other architectures, including a large language model, and found that GNNs more accurately identify active, drug-like molecules that are structurally distinct from the training set and known antibiotics. Using the model to virtually screen ~6 million compounds, we identified 213 compounds for experimental validation and found that 83 (39%) inhibited N. gonorrhoeae growth. Two of these compounds were structurally dissimilar to existing antibiotics, maintained potency against multidrug-resistant N. gonorrhoeae strains in vitro, exhibited promising selectivity indices, and were rapidly bactericidal with low frequencies of resistance. Proteomic studies revealed their distinct mechanisms of action, with one compound targeting alanine racemase, an enzyme involved in the essential process of peptidoglycan synthesis. Furthermore, the compounds showed early promise in reducing N. gonorrhoeae titers in a human vagina-on-a-chip infection model and a mouse vaginal infection model. Our work establishes the deep learning–enabled discovery of selective antibacterial compounds against N. gonorrhoeae as a much-needed hit discovery tool to address the growing crisis of antimicrobial resistance for this pathogen.
|
|
Scooped by
mhryu@live.com
Today, 2:40 PM
|
Antimicrobial peptides (AMP) are essential components of the innate immune system in animals, playing crucial roles in defending against microbial infections. Hydrogen sulfide (H2S), a vital gasotransmitter, is involved in regulating a wide array of physiological functions. However, the role of H₂S in the anti-infective actions of AMPs remains poorly understood. Here, we demonstrate the crucial role of H2S in mediating the anti-infective functions of AMPs across diverse animal species. LL-37/CRAMP, the human cathelicidin family AMP and its mouse homolog, induce H2S production predominantly through 3-mercaptopyruvate sulfurtransferase (3MST) in E. coli and Clostridium perfringens. The generated H₂S further participates in reactive oxygen species (ROS) accumulation, leading to bacterial death by oxidative damage. Additionally, LL-37 activates the Akt signaling pathway, upregulating cystathionine γ-lyase (CSE) expression to generate H2S in macrophages. This process plays a potential role in facilitating the immunomodulatory activity of LL-37/CRAMP and we confirmed that H2S is involved in their in vivo anti-infective activity of LL-37. Moreover, besides LL-37/CRAMP, we reveal that AMPs from various animal species also induce H2S production in bacteria and macrophages. Overall, our findings highlight a previously unrecognized role of H2S in AMP anti-infective function, which is ubiquitous across AMPs from different animals. In this work, authors show that LL-37/CRAMP induces hydrogen sulfide production in bacteria and macrophages. The generated H₂S contributes to the antimicrobial and immunomodulatory functions of LL-37/CRAMP, which is ubiquitous across AMPs.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
All kingdoms of life have developed strategies to limit viral infection. In humans, interferons induce a suite of antiviral factors that collectively provide immunity. Some human immune genes are homologous with antiphage defense genes in bacteria, though the extent of this overlap is not known. Here, we screened a panel of human innate immune genes for phage defense in E. coli and found that the RNA exonuclease ISG20 potently restricts the RNA phages MS2 and Qβ. Purified ISG20 trims the 3-prime untranslated regions (UTRs) of RNA phage genomes, explaining its ability to block phage replication in E. coli. Homologs of ISG20 from bacteria function similarly, exhibiting nuclease-dependent antiphage defense in bacteria and UTR-trimming in vitro. When expressed in human cells, these bacterial exonucleases also restrict human RNA viruses with similar potency as the human antiviral protein ISG20. Thus, antiviral genes from both humans and bacteria can function interchangeably. This bidirectional, interkingdom immunity suggests that viral targets overlap, implying that both bacterial and human factors recognize ancient features of RNA viruses.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
Many antibiotics are ineffective against the Gram-negative pathogen Pseudomonas aeruginosa because of intrinsic defence mechanisms, such as the impermeable bacterial outer membrane. Here, we show that protein antibiotics called L-type pyocins kill P. aeruginosa by inhibiting the β-barrel assembly machinery (BAM) complex at the cell surface, halting outer-membrane protein assembly. Using single-particle cryo-electron microscopy, we show that L-type pyocins bind a surface-exposed region of BamA and deploy a C-terminal peptide that competitively inhibits the BAM complex, demonstrating that cell entry is not required for antibiotic activity. We combine genetics, multi-omics and cryo-electron tomography to show that BAM complex inhibition by L-type pyocins or the cyclic-peptide antibiotic, darobactin, triggers a multifaceted transcriptomic, proteomic, and morphological response. BAM inhibition ultimately leads to a catastrophic loss of membrane integrity and cell death. These results validate BAM as a target for antibiotics that do not enter the cell and define an engineerable system for their development. L-type pyocins are a class of bacteriocin-like proteins with antibacterial activity. Here, the authors show that these proteins kill the pathogen Pseudomonas aeruginosa by inhibiting the BAM complex at the bacterial cell surface, halting outer-membrane protein assembly.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
Cell differentiation and Turing-like patterning are tightly associated in a group of filamentous cyanobacteria that differentiate specialized N2-fixing cells, called heterocysts. Based on systematic genetic analyses, in particular genome-wide identification of recognized promoters and assays with a reconstituted Anabaena transcription system in E. coli, we established HetZ as the central activator of the gene regulatory network of heterocyst differentiation. Biochemical and cryo-EM analyses further established HetZ as a σ factor (σHetZ). Unique domain insertions in σHetZ are involved in promoter DNA recognition-unwinding and interaction with the inhibitor PatU3. σHetZ-PatU3 and the master regulator-diffusible inhibitor constitute the minimal core regulatory circuit (CRC) for cell fate determination and patterning. σHetZ activates not only genes of the CRC but also downstream regulator/effector genes involved in morphological and functional development. The gene regulatory network and the structure–function relationship of σHetZ depict how cell differentiation and patterning are coordinated in this group of multicellular cyanobacteria.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Agricultural fields are increasingly contaminated with aromatic ester compounds originating from insecticides, herbicides, plastic mulching films, adjuvants and stabilizers used in agrochemicals. Many of these esters are persistent and bioaccumulate in the food chain, posing serious risks to non-target biota, including crops and humans. Among various remediation strategies available, bioremediation represents a cost-effective, sustainable, and eco-friendly approach for the clean-up of these contaminants. This review describes types of aromatic ester (aromatic-acid and aromatic-alcohol esters) pollutants and their bacterial degradation. The recent advances in understanding genetic, metabolic, evolutionary aspects of bacteria and synthetic biology approaches that aid in degradation of these xenobiotics are discussed. Additionally, it provides insights into the assistive eco-physiological traits of aromatic ester-degrading bacteria, which collectively enhance in situ degradation and offer promising avenues for sustainable agricultural practices and restoration of contaminated agro-ecosystems.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
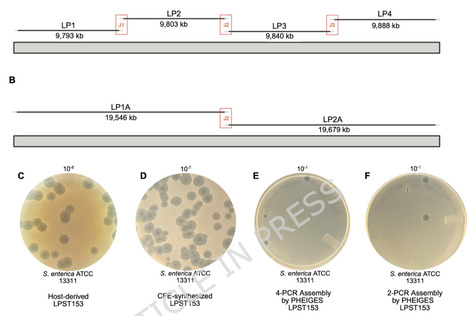
Cell-free gene expression (CFE) systems provide a rapid and modular platform for synthesizing bacteriophages without the need for living host cells. However, the generalizability of CFE-based phage synthesis across diverse phages, as well as the functional comparability of in vitro-synthesized phages to host-derived counterparts, has not yet been fully explored. In this study, we evaluated the ability of an E. coli-based CFE platform to synthesize diverse bacteriophages relevant to food safety and synthetic biology applications. Using an E. coli–based CFE system, we synthesized seven phages from four different families, achieving infectious titers ranging from 10⁶ to 10¹¹ plaque-forming units per milliliter (PFU/mL). Of these seven phages, five phages, vB_SalM-LPST153 (LPST153), vB_SenM-S16 (S16), SP6, vB_Sens_Jbel (Jbel), and vB_EcoM_Alf5 (Alf5), are reported here for the first time as successfully synthesized and characterized in a CFE system. CFE-synthesized phages remained capable of infecting host strains; however, significant differences in efficiency of plating (EOP) were observed across multiple Salmonella, E. coli, and Shigella hosts. Long-term storage tests for LPST153 showed that phage buffer (PB) stably maintained CFE titers above 10⁹ PFU/mL for 12 weeks, while purified CFE-synthesized and host-derived LPST153 lysates exhibited broadly comparable thermal stability profiles across the tested temperatures. Using the PHEIGES workflow, we also demonstrated complete in vitro assembly and expression of LPST153 from PCR fragments. In addition, phages were successfully synthesized using CFE lysates derived from the endotoxin-free E. coli strain ClearColi™, demonstrating the synthesis of infectious phages in an endotoxin-free system. These findings expand the diversity of bacteriophages compatible with cell-free synthesis (CFS) and demonstrate that CFE-synthesized phages remain infectious and retain several functional properties, although some differences in host-dependent infectivity were observed. The ability to synthesize infectious phages in endotoxin-free systems and assemble phage genomes entirely in vitro highlights the potential of CFE platforms for scalable, decentralized, and cold–chain–independent manufacturing of phage-based therapeutics and food safety interventions.
|
|
Scooped by
mhryu@live.com
Today, 12:28 AM
|
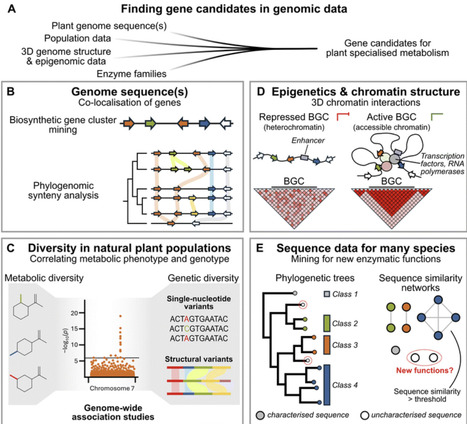
Rapid technological progress has enabled plant biologists to accumulate unprecedented volumes of multi-scale, multi-modal data, yet this abundance of data has intensified the challenge of translating complexity into biological understanding. Foundation models (FMs), large-scale artificial intelligence (AI) systems pretrained on millions of sequences, structures, or images and adaptable to diverse tasks are breaking through this barrier. Across plant science, these FMs are already making an impact: genomic FMs decode regulatory grammar, protein FMs enable rational protein engineering, vision FMs score phenotypes at breeding-population scale, single-cell FMs annotate cell types across species, and FM-powered AI agents accelerate knowledge retrieval and automate research workflows. While experimental validation remains indispensable, foundation models empower plant scientists to accelerate scientific discovery.
|
|
Scooped by
mhryu@live.com
Today, 12:05 AM
|
Intracellular bacterial symbioses have arisen myriad times in eukaryotes, with dozens known from insects alone. Beginning with Buchnera, the obligate endosymbiont of aphids, genomes of endosymbionts have illuminated their evolutionary origins and metabolic contributions to hosts. However, the mechanisms by which non-culturable endosymbionts enter host cells and suppress cellular immune processes have remained unclear. Here we show that an uncharacterized Buchnera protein, designated SyeA, was present in the Buchnera ancestor, is secreted into the host cytoplasm, is homologous to secreted effectors of bacterial pathogens and is essential for Buchnera transmission. Buchnera is transmitted through expulsion from specialized maternal cells and uptake by embryos. Using immunofluorescence microscopy, we found elevated SyeA levels after colonization of the embryonic cell, accompanied by actin accumulation at the entry site. SyeA localizes outside the host-derived membrane and actin layer surrounding each Buchnera cell within host cytoplasm. Knockdown of syeA expression disrupts colonization of embryos and embryonic development and elevates lysosomal activity, leading to Buchnera destruction. Our findings provide insights into how an anciently associated, mutualistic endosymbiont achieves its intracellular existence. SyeA represents a vestige of pathogenic origins that was followed by evolution of increased host control and erosion of the original, more complex pathogenicity machinery. SyeA was present in the Buchnera ancestor, is secreted into the host cytoplasm, is homologous to secreted effectors of bacterial pathogens and is essential for Buchnera transmission.
|
|
|
Scooped by
mhryu@live.com
Today, 5:13 PM
|
Current cytosine base editors (CBEs) are limited to unidirectional C to T conversions, restricting their applications. Retrons, bacterial genetic elements, encode a reverse transcriptase that generates multicopy single-stranded DNA (msDNA) by reverse transcribing specific non-coding RNA (ncRNA). This msDNA mimics Okazaki fragments during DNA replication, making retrons promising for gene editing. Here, we developed a retron-based cytosine base editor (RCBE) by fusing cytosine deaminase with reverse transcriptase (RT-CDA) within the retron system. RCBE first transcribes ncRNA, allowing RT-CDA to deaminate cytosine on the ncRNA. The modified ncRNA is then reverse transcribed into msDNA, where RT-CDA induces further cytosine deamination. This mutant msDNA introduces specific mutations into target gene sequences, enabling both C to T and G to A conversions. Using RCBE, we demonstrated accelerated molecular evolution of the rpoB gene in E. coli. High-throughput sequencing confirmed that RCBE achieves a mutation rate of up to 0.2% in regions with high GC content. Our findings establish RCBE as a versatile tool, particularly suitable for directed evolution in GC-rich regions, with broad potential applications across various bacterial and eukaryotic hosts.
|
|
Scooped by
mhryu@live.com
Today, 4:13 PM
|
Synthetic gene circuits provide experimentally tractable systems for dissecting how genetic feedback and diffusible signals generate multicellular patterns. However, building multi-component circuits whose behavior can be quantitatively linked to module-level measurements, diffusion, and spatial boundary conditions remains challenging. Here, we designed and engineered a bacterial patterning system in which positive feedback, delayed negative feedback, and two orthogonal quorum-sensing signals are integrated in E. coli. We first implemented and characterized the feedback modules separately, measured the effective diffusion of the signals in the experimental setup, and used these data to parameterize a mathematical model. In quasi-2D bacterial lawns, the complete circuit generated self-organized spatiotemporal dynamics consisting of an sfGFP activation front followed by successive mCherry propagating pulses/traveling waves. Model-guided perturbations showed that lawn size, lawn position relative to the domain boundary, and signal degradation modulate the timing, amplitude, wavelength, and directionality of these patterns. Our work establishes a modular synthetic multicellular reaction-diffusion system in which circuit architecture, signal diffusion, and boundary-mediated signal exchange can be experimentally connected to emergent patterning dynamics.
|
|
Scooped by
mhryu@live.com
Today, 3:51 PM
|
How microbial populations respond to repeated environmental transitions determines both their ecological fitness and their utility in biotechnological applications. Using Pseudomonas putida KT2440 equipped with fluorescent biosensors and monitored by automated flow cytometry in the Segregostat platform, we show that exposure to benzoate, a plastic-derived aromatic feedstock, progressively reduces cellular responsiveness, defined as the fraction of cells that successfully activate a gene circuit following an environmental transition. Unlike classical switching costs, which promote phenotypic diversification, benzoate suppresses responsiveness without increasing population entropy, in a concentration-dependent and circuit-independent manner tightly correlated with fitness loss. A resource allocation model incorporating the competing demands of benzoate assimilation, toxicity, and tolerance reveals that this impairment emerges from a three-way competition for limited cellular resources. Above a critical benzoate load, insufficient resources remain available to sustain the adaptive reallocation required for circuit activation. In continuous culture, a non-responsive subpopulation accumulates as a leading indicator of population collapse. Exploiting this signal, we implement a two-stage connected bioreactor system in which benzoate feeding is autonomously regulated based on real-time population structure, enabling complete substrate consumption and stable operation at otherwise destabilizing concentrations. These results establish cellular responsiveness as a quantitative population variable and demonstrate that structure-aware feedback control, acting on population composition rather than bulk physiology, provides a principled route toward autonomous bioprocesses on challenging substrates.
|
|
Scooped by
mhryu@live.com
Today, 3:03 PM
|
Non-coding RNAs play diverse roles in a wide range of cellular processes, with their spatial structure being pivotal to their function. RNA secondary structure is a key determinant of its overall fold. Given the scarcity of experimentally determined RNA 3D structures, understanding secondary structure is vital for discerning RNA function. Currently, there is no universally effective solution for de novo RNA secondary structure prediction. Existing methods are becoming increasingly complex without marked improvements in accuracy and often overlook critical features such as pseudoknots and alternative folds. Here, we introduce SQUARNA, a new approach to de novo RNA secondary structure prediction that is suitable for both individual RNA analysis and large-scale structural searches. SQUARNA revisits the concept of base pair maximization and develops it into a stem maximization idea coupled with the widely used free energy minimization (MFE) framework. SQUARNA can predict alternative structures and handle pseudoknots of arbitrary complexity. Benchmarking shows that SQUARNA outperforms existing methods, including deep learning models, in both single-sequence and alignment-based RNA secondary structure prediction. SQUARNA seamlessly integrates sequence and alignment information with experimental data, such as residue reactivities obtained by chemical probing, as well as other structural restraints, including automated searches for Rfam database templates, G-quadruplex patterns, and protein-binding motifs. SQUARNA is available as a standalone tool at https://github.com/febos/SQUARNA and as a web server at https://larnal.imol.institute.
|
|
Scooped by
mhryu@live.com
Today, 2:44 PM
|
Akkermansia muciniphila is a key member of the gut microbiota and plays important roles in host metabolism and health. In the colon, A. muciniphila extracts nutrients from oligosaccharide-rich mucin glycans that comprise the mucosa. However, this environment is complex and shaped by dietary inputs, microbiome metabolism, and mucin glycan composition varying across hosts, gastrointestinal regions, and physiological states. How strains of A. muciniphila integrate these nutrient signals into growth initiation and niche colonization remains unclear. Here, we compare physiological responses of a human- and mouse-derived strain of A. muciniphila, finding that dietary sugars differentially affect these isolates, suggesting host-associated tuning of metabolic capacity. In contrast, several mucin-derived sugars impose a conserved, concentration-dependent delay in growth initiation, implicating the lag phase as a critical metabolic checkpoint for growth. Genetic suppressor analysis identified sugar kinases and a component of the tricarboxylic acid cycle as genetically encoded control points linking glycan sugar exposure to the energy balance required for growth. These findings demonstrate that mucin-derived sugars function as both nutrients and metabolic stressors, regulating growth initiation. We propose that A. muciniphila employs metabolic “brakes” to coordinate growth with mucin composition, putatively linking host glycan landscapes to microbial physiology and ecological fitness within the mucus layer.
|
|
Scooped by
mhryu@live.com
Today, 2:28 PM
|
Optimizing growth conditions and culture media is a major goal in microbiology. A challenge is that nutrients can have complex, non-additive effects on growth. The fact that resources interact with one another has long been known in ecology, but systematic maps of resource interactions at all orders have been lacking and it is not known whether interactions are primarily low order or fundamentally complex. To tackle this problem, we have followed a full factorial design approach and measured the growth of seven different bacterial species in all possible combinations of 8 carbon sources under carbon-limiting conditions. Our approach allows us to directly estimate interactions at all orders. Even though all C-sources stimulate growth on their own as well as in combination with other nutrients, most of them can also have negative effects on growth when they are added to at least some nutrient mixtures. We show that the switch from positive to negative fitness effects is governed by global epistasis among resources. An analysis of variance shows that additive effects and pairwise interactions explain most of the variation in fitness, allowing us to train simple regression models that accurately predict bacterial growth in novel environments. The generality of these findings across seven different bacterial strains belonging to two different families indicates that interactions between carbon sources under carbon-limiting conditions may be generally learnable from a relatively sparse set of constructed environments, enabling the rational optimization of growth conditions.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
Schlafen nucleases restrict viral infection in mammals by cleaving self RNAs, however, their function and mechanism in prokaryotic immunity is unknown. Here, we uncover CRISPR-associated Schlafen (Cash) proteins containing a Schlafen domain fused to Csx15, an uncharacterized member of Rossmann-like nucleotide-binding sensors. Cash is activated by cyclic tetra-adenylate (cA₄) produced during type III CRISPR interference and induces cell toxicity by cleaving tRNAs, primarily in the T-loop. Cryo-electron microscopy structures of Chloroflexi bacterium Cash reveal an inactive dodecamer, the formation of a filament upon cA₄ binding to align catalytic interfaces, and the molecular basis of substrate recognition and cleavage in a tRNA–bound complex. We identify numerous families of prokaryotic Schlafen proteins associated with diverse antiviral defense systems and characterized by unique sensor domains. This work highlights tRNA depletion by Schlafen nucleases as an evolutionary recurring antiviral strategy and reveals mechanistic differences between Cash and human Schlafen members. Schlafen nucleases restrict viral infection in mammals by cleaving self RNAs. Here, the authors identify bacterial Schlafen nucleases as components of anti-phage type III CRISPR systems that restrict viral infection through tRNA depletion.
|
|
Scooped by
mhryu@live.com
Today, 1:00 AM
|
E. coli encounters chemically diverse carbon sources, and the observed outputs of its transcriptional regulatory network (TRN) vary with substrate chemistry, metabolic entry route, and growth physiology. Here, we compiled PRECISE-NP881, an 881-condition transcriptome compendium comprising 346 RNA-seq profiles generated for this study during growth on 43 individual carbon sources, and used independent component analysis to quantify condition-specific activities of 137 iModulons, defined here as statistically independent gene-expression modules. We identified 25 carbon-catabolism iModulons and summarized their activity patterns across the 43 substrates into four activity-defined substrate groups. These activity patterns were associated with measured growth rates, substrate chemical classes, central-metabolic entry routes, carbon-normalized stoichiometric yield, and model-estimated proteome allocation. Faster-growing sugar conditions showed low CRP-linked iModulon activity, whereas slower-growing conditions showed elevated, condition-specific activity of CRP-linked and substrate-specific catabolic iModulons. TCA-entry and amino acid–associated conditions were linked with NtrC-1 and Propionate iModulon activities, with targeted knock-out assays supporting the conditional physiological relevance of selected propionyl-CoA-associated genes. A subset of nitrogen-containing, slower-growth conditions with predicted ammonium release induced the cryptic prophage-associated SgcABCEQX iModulon. Projection of an independent glucose starvation/refeeding time-course dataset revealed overlapping dynamics among selected carbon-catabolism iModulons and coordinated changes in growth- and stress-associated TRN outputs. Together, these results provide a systems-level atlas of observed carbon-responsive transcriptional states and systematize carbon physiology at scale.
|
|
Scooped by
mhryu@live.com
Today, 12:56 AM
|
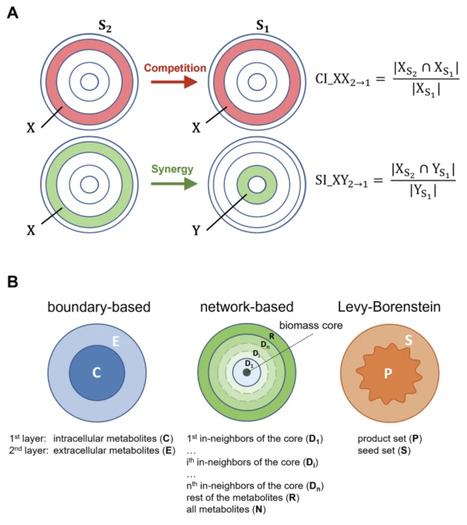
Understanding the composition of microbial communities in their environment remains a challenge due to the complex interplay of factors like inter-species interactions and nutrient availability. In this context, it has become an established approach to use overlap in functional subsets of metabolic networks as indices of synergy and competition among microorganisms. Here, we show that this idea can actually be reduced to a much simpler principle. Leveraging the agent-based community modeling software BacArena and natural co-occurrence patterns in the human gut microbiome for a systematic comparison, we find that simple set-theoretical indices explain interactions to a similarly high degree as more sophisticated, established approaches based on network topology. Furthermore, we observe that the performance of most indices decreases substantially for patients diagnosed with obesity or inflammatory bowel disease, suggesting a systemic decline in the microbiome.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
Isoprenoid quinones are redox-active lipids essential for numerous cellular processes, including ATP synthesis via electron transport chains. They are found across all domains of life. Their diversity among microbial taxa has established them as chemotaxonomic markers, while recent studies have linked quinone repertoire variations to metabolic adaptations, and have identified novel quinone types. Despite this renewed interest, a comprehensive overview of quinone distribution across Bacteria is missing. Here, we systematically annotated quinone biosynthetic pathways for major quinones (ubiquinone, rhodoquinone, plastoquinone, and menaquinone) across 26,264 high-quality bacterial genomes. We also text-mined quinone data from over 6,000 species in published abstracts to validate our genomic annotations while compiling valuable information on quinone chains. Mapping these data onto a phylogenetic framework provides the most extensive synthesis of quinone distribution in Bacteria to date. Our analysis reveals an unexpectedly dynamic evolutionary history for the two menaquinone-producing pathways. Furthermore, the discovery and experimental validation of a deeply branching ubiquinone pathway in the phylum Desulfobacterota provide the first evidence of such a pathway outside the phylum Pseudomonadota, offering new insights into the nature of the ancestral ubiquinone biosynthetic route. Beyond these discoveries, the global overview of bacterial quinones distribution presented here paves the way for future investigations into quinone-associated biochemistry and physiology, including predicting quinone structural features from genomic data, exploring correlations between quinone structures and cellular traits, and studying quinone repertoire evolution in relation to microbial metabolic diversification.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
Poly(ethylene terephthalate) (PET) is one of the most widely used plastics in food and textile applications, yet post-consumer PET waste persists and accumulates in the environment as macro- and microplastics with adverse health and ecological impacts. Although PET-hydrolysing enzymes have been extensively studied in vitro, whole-cell microbial systems capable of using PET as a growth substrate remain limited, particularly for difficult-to-collect waste. Here, we engineer an environmental bacterium to directly assimilate PET. A strain of Pseudomonas umsongensis capable of metabolizing the PET monomer terephthalic acid was isolated and engineered to secrete the high-activity PET hydrolase polyester hydrolase Leipzig 7 via a recombinant twin-arginine translocation motif signal peptide. PET bioavailability was further enhanced through solvent-based pretreatment to generate an amorphous, macroporous substrate. The engineered strain demonstrated direct PET utilisation and hydrolysis, supporting self-sustaining microbial growth. Additionally, in non-sterile wastewater the strain survived and hydrolysed PET microplastics, highlighting its potential for bioremediation and sustainable upcycling applications.
|
|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
To advance the computational simulation of cellular life, we propose a virtual yeast, an artificial intelligence (AI)-driven agent that models eukaryotic cellular behaviors by integrating multimodal biological data, mechanistic reasoning and active experimentation using Saccharomyces cerevisiae as a genetically tractable and data-rich model system. Cellular complexity is decomposed into eight function-centred modules, spanning genetic, metabolic and structural systems, each realized as a domain-specific AI tool coordinated through a large language model-based orchestration layer. Built on three data pillars, namely, mechanistic knowledge, subcellular architecture and dynamic states, the system integrates representation learning and generative modelling within a closed-loop learning pipeline that autonomously designs and executes experiments. The virtual yeast serves as both a conceptual and an operational platform to optimize biosynthetic pathways, support the generation and prioritization of hypotheses across diverse cellular processes, and accelerate target discovery. By coupling biological realism with autonomous AI reasoning, the virtual yeast establishes a generalizable blueprint for constructing virtual eukaryotic cells and advancing synthetic biology. A virtual yeast, an artificial intelligence-driven agent for modelling eukaryotic cellular behaviours, is described.
|































1str, bgc