 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:10 AM
|
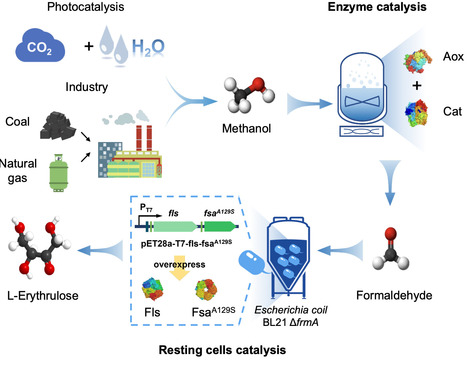
The biocatalytic conversion of green methanol, a promising feedstock, into high value-added products is seen as an attractive way to establish sustainable biomanufacturing. Although natural and engineered microorganisms can utilize methanol, efficiently converting C1 carbon into valuable chemicals in growing cells remains challenging because non-native pathways often suffer from low carbon utilization and limited catalytic stability. Here, we report the integration of an engineered enzyme cascade into resting cells and its coupling with a screened alcohol oxidase enzyme to develop an efficient and stable enzyme-resting cell cascade system for methanol conversion, achieving customizable production of L-erythrulose (C4 sugar) or L-sorbose (C6 sugar). This system shows a stimulated carbon atom economy for L-erythrulose production from methanol and significantly improved stability compared to free enzyme catalysis. By integrating enzyme cascades with durable resting cells, this strategy provides a versatile platform for converting methanol and other C1 substrates into value-added products. Methanol use in engineered methylotrophs is constrained by toxicity and low efficiency. Here, the authors report the engineering E. coli resting cells coupled with a screened alcohol oxidase to enable biocatalytic production of rare sugars from methanol as the sole substrate.
|
|
Scooped by
mhryu@live.com
July 18, 11:45 PM
|
With this status report, we aim to provide a timely snapshot of the protein engineering field as a broad and rapidly advancing discipline that integrates computational, molecular biology, structure-guided, evolutionary, and synthetic approaches to create new and improved proteins with tailored structures and useful functions. The report is organized into eight thematic areas spanning core methodologies and major application domains, including enzymes, therapeutics, detection, synthetic biology, and materials. Contributions from experts across these areas highlight both the historical foundations and recent advances in their respective fields, with particular emphasis on the growing influence of machine learning and artificial intelligence-based methods. Emerging from this broad overview is a central message: protein engineering appears to be entering a golden age, defined by a rapidly accelerating pace of progress, even as significant challenges in design, screening, and real-world application remain. Looking ahead, the continued integration of computational and experimental strategies is poised to further accelerate the impact of protein engineering across an expanding range of economically and societally important sectors, from therapeutics and molecular imaging to diagnostics, plastic recycling, and industrial chemistry.
|
|
Scooped by
mhryu@live.com
July 18, 4:59 PM
|
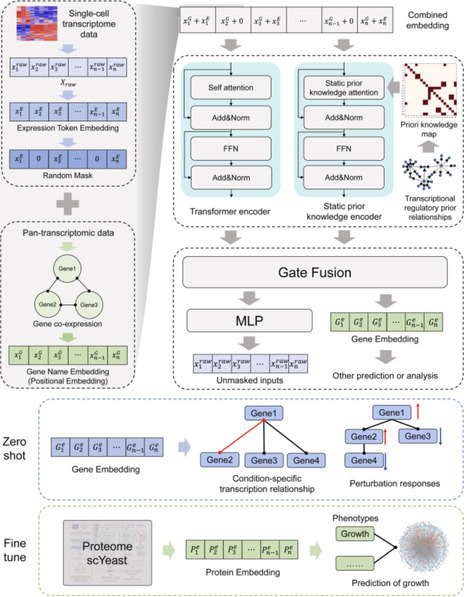
Though large-scale pre-trained models are vital for foundational cell modeling, most of them focus on human or mouse systems, with less emphasis on model organisms like yeast (Saccharomyces cerevisiae), and fail to use existing biological prior knowledge effectively. Here, we present scYeast, the first foundational cell model for yeast single-cell transcriptomics that effectively embeds biological priors. scYeast employs a novel asymmetric parallel architecture to infuse transcriptional regulatory information into the Transformer's attention mechanism, leveraging biological knowledge during training. Pre-trained on large-scale yeast single-cell transcriptomics data, scYeast demonstrates strong generalization and biological interpretability. It shows capability in zero-shot tasks, such as inferring regulatory relationships. After fine-tuning, scYeast performs well in diverse tasks, including cell state classification, growth doubling time prediction, and gene perturbation response prediction. Additionally, using transfer learning, scYeast can be adapted to other omics datasets, such as proteomics, thus broadening its utility. Overall, scYeast is a promising tool for yeast single-cell biology research and presents a new framework for integrating foundational models with biological priors, accelerating discovery in yeast synthetic and systems biology and providing a replicable framework for other organisms.
|
|
Scooped by
mhryu@live.com
July 18, 4:53 PM
|
Arsenic contamination of drinking water remains a persistent global health burden and an environmental justice challenge, particularly for low-resource communities that lack access to reliable monitoring tools. Synthetic-biology-driven biosensors offer a promising complement to conventional analytical methods by coupling arsenic-responsive genetic circuits with portable, low-cost readouts suitable for field deployment. This review traces the evolution from the early ArsR-based E. coli sensor to modern whole-cell and cell-free platforms that approach World Health Organization–relevant detection limits for arsenic in water under controlled conditions, emphasizing how signal amplification strategies intersect with shelf life, biosafety, and regulatory simplicity. The operational principles of ars operon–derived modules are examined across detection, processing, and host-engineering layers that collectively tune sensitivity, dynamic range, and robustness. Immobilization formats, microfluidic architectures, and transduction mechanisms─including colorimetric, fluorescent, bioluminescent, and electrochemical outputs─are analyzed for their ability to integrate biological sensing with commodity optics and electronics in portable devices. Building on this engineering landscape, the review highlights how biodesign automation, high-throughput Design–Build–Test–Learn workflows, and emerging AI tools such as supervised learning and Bayesian optimization are accelerating the construction and optimization of arsenic-responsive genetic circuits. Biosafety and regulatory considerations, including biocontainment, standardized stress-testing, and community codesign, are discussed to position arsenic biosensors as candidates for integration into distributed water-quality monitoring networks that combine synthetic biology, low-cost hardware, automation, and AI under robust governance regimes.
|
|
Scooped by
mhryu@live.com
July 18, 4:38 PM
|
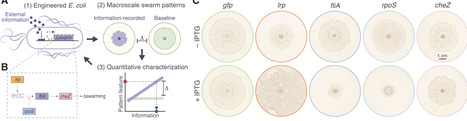
Many bacterial species form self-organized macroscale patterns through swarming. Despite its extensive genetic tractability, Escherichia coli remains underexplored for robust, applied control of swarming. Here we develop a set of E. coli strains that generate centimeter-scale swarming patterns to spatially record environmental inputs. Specifically, we modulate the expression of swarming-related genes in response to chemical and optical signals, reshaping baseline swarm patterns in analog or binary-like fashions. To decode bacterial patterns across space and time, we develop scalable computational methods incorporating feature extraction, regression, and deep-learning models. Time-lapse imaging reveals that colonies record inputs dynamically, enabling early-stage classification. This work establishes a strategy for spatial information recording in E. coli and expands the toolkit for programming emergent microbial behaviors at macroscopic scales.
|
|
Scooped by
mhryu@live.com
July 18, 4:31 PM
|
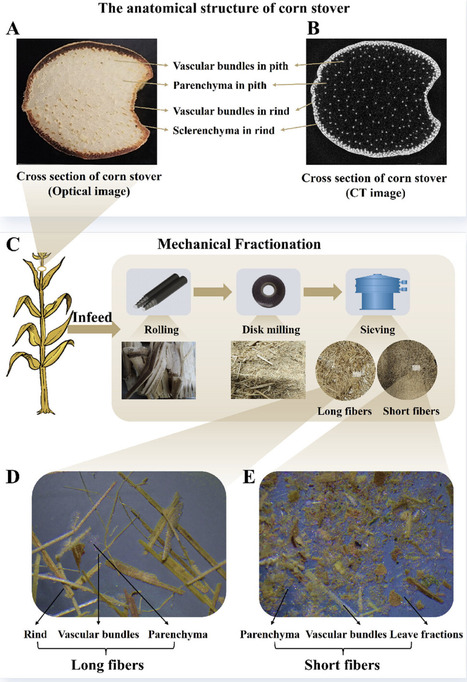
Lignocellulosic biorefinery constitutes a critical pillar for transitioning the fossil-based industrial paradigm toward sustainability. However, in lignocellulosic biorefinery, cross-interference between cellulose, hemicellulose, and lignin persists throughout all steps. Effective regulation must extend beyond pretreatment across the entire process. Here, we develop a whole-process regulation strategy for corn stover. Mechanical fractionation homogenizes physical structure, yielding parenchyma-rich short fibers and vascular-bundle-dominant long fibers. For highly degradable short fibers, molecular control by methanol during steam explosion suppresses lignin condensation, followed by oxidative enhancement by carbon quantum dots during enzymatic hydrolysis, boosting cellulose conversion and facilitating mild lignin depolymerization for high-performance epoxy resins. For high-crystallinity long fibers, two-stage selective enzymatic hydrolysis preserves crystallinity to produce cellulose nanocrystals. Techno-economic analysis shows a 36.7% revenue increase over the unregulated baseline. This integrated approach embodies the concept of precision biorefinery: a transformative framework where whole-process regulation orchestrates multi-level heterogeneity-guided fractionation to enable full-component directed valorization, ensuring compatibility between biomass attributes, process, and product specifications. The concept and innovations have been industrially validated.
|
|
Scooped by
mhryu@live.com
July 18, 4:19 PM
|
Infectious diseases continue to pose unprecedented challenges to public health and the global economy. Virulence factors (VFs) enable pathogens to adhere, reproduce, and cause damage to host cells, while antibiotic resistance genes (ARGs) enable pathogens to withstand treatments that would otherwise be effective. The concurrent identification of VFs and ARGs is crucial for efficient pathogen surveillance. However, existing tools for predicting VFs or ARGs typically suffer from high false negative rates and limitations in identifying only high-identity genes against known reference VF or ARG databases. To address these challenges, we developed SEVA, an advanced model that integrates protein language models (pLMs) with structural and evolutionary protein features to predict VFs and ARGs from genome sequencing data. Integrating multiple homologous sequences can identify latent virulence or drug resistance caused by site mutations, reducing false negative rates. Meanwhile, the protein structure remains conserved despite the low sequence identity in some functional domains of VFs or ARGs. The aggregate of protein structure information further improves the identification abilities of VF and ARG. In addition, pLMs enable the model to capture high-dimensional feature representations more effectively. SEVA rigorously collected three datasets with over 20,000 genes and five reference databases. It outperforms state-of-the-art methods, including Diamond, VRprofile, FoldSeek, PreVFs-RG, PLM-ARG, ARG-BERT, and HyperVR, achieving an accuracy of 97.13% and confirming the efficacy of its key components, such as refined feature selection and multiple sequence alignment subsampling. SEVA takes protein sequences as input and derives evolutionary, structural, and statistical representations for prediction, making our model a reliable tool for VF and ARG prediction. This capability is particularly valuable in epidemic prevention and control, where accurate identification of VFs and ARGs is crucial. By providing concurrent and reliable predictions of VFs and ARGs, SEVA enhances our ability to respond to microbial threats effectively. This finding supports robust efforts to mitigate the spread of infectious diseases and safeguard public health, addressing a critical gap in contemporary epidemic response strategies.
|
|
Scooped by
mhryu@live.com
July 18, 3:06 PM
|
The proliferation of pathogen bioinformatics pipelines has outpaced the community ability to compare them on common ground. Self-reported performance numbers, ad-hoc evaluation datasets, and inconsistent metrics make pipeline selection difficult for clinical and public-health researchers. We present PathoBench, an open web platform that addresses this gap through three coordinated mechanisms: (i) a curated registry of 26 standard benchmark datasets across 10 human pathogens, each with persistent identifiers and direct download links; (ii) pathogen-specific evaluation metrics that submissions must report, allowing direct head-to-head comparison only on the same dataset; and (iii) a credibility framework combining mandatory dataset attestation, ORCID-linked attribution, public peer comments, and administrator verification. As a case study, four published Mycobacterium tuberculosis drug-resistance pipelines were evaluated against the WHO TB mutation catalogue, demonstrating the framework discriminating power. PathoBench is open for community contributions across all ten supported pathogens.
|
|
Scooped by
mhryu@live.com
July 18, 11:02 AM
|
The regulation of protein stability is essential for cellular homeostasis and is determined by a combination of intrinsic sequence motifs and extrinsic recognition enzymes. Despite growing knowledge of the protein degradation machinery, the ability to predict a protein's stability from its amino acid sequence remains challenging. Here we develop a machine learning model to predict protein stability from N-terminal amino acid sequences. Using our model and experimental validation, we identify known and novel sequence motifs governing protein stability. We additionally use this model to predict the stability of alternative translational isoforms with distinct N-termini produced from the same mRNA. Despite differing by a limited number of amino acids, we identify N-terminal isoforms with drastically different stabilities relative to their annotated counterparts, highlighting the potential of N-terminal extensions and truncations to regulate protein function. Together, this model provides a valuable tool for evaluating additional protein datasets and protein design strategies.
|
|
Scooped by
mhryu@live.com
July 18, 10:49 AM
|
Bacteria, as simple unicellular microorganisms, can directly lyse tumor cells following intratumoral administration. Flagellin and cell wall components derived from bacteria can reshape the tumor microenvironment and activate host anti-tumor immune responses. Notably, certain anaerobic and facultative anaerobic bacteria exhibits preferential colonization in the hypoxic core of solid tumors, where they can serve as in situ “micro-bioreactors” and targeted delivery vectors for synergistic tumor therapy. Based on rapid advances in synthetic biology, diverse engineered bacteria and bacteria-based biohybrid systems have been developed for precision tumor therapy, including gene restoration, environment-responsive programmable expression, cytotoxic protein production, and multimodal combination therapy with nanomaterials, immune modulators, or photothermal agents. These living platforms enable targeted drug/gene delivery, sustained local therapeutic output, and durable anti-tumor immune activation. However, clinical translation remains hindered by insufficient biosafety control, off-target colonization, uncontrolled proliferation, immune clearance, and lack of standardized manufacturing and regulatory guidelines. This review systematically summarizes the historical development, inherent anti-tumor mechanisms, genetic engineering strategies, and multimodal biohybrid systems of bacteria-mediated tumor therapy. We comprehensively discuss the advantages, limitations, comparative characteristics, and core bottlenecks of different platforms, and propose rational design strategies for next-generation intelligent, controllable, and clinically translatable bacteria-derived living anti-tumor systems.
|
|
Scooped by
mhryu@live.com
July 18, 10:41 AM
|
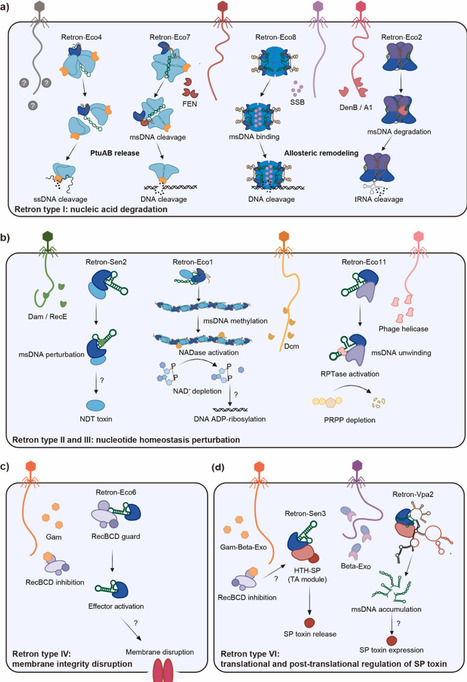
Bacteria employ a sophisticated arsenal of immune systems to counter bacteriophage infection. Among these systems, retron antiphage systems have emerged as a widespread class of reverse transcriptase (RT)-associated regulatory modules. Retrons are genetic elements encoding an RT and a structured noncoding RNA (ncRNA), which together generate multicopy single-stranded DNA (msDNA). In antiphage defense, an RT-msDNA module associates with its cognate effector protein to form a tripartite complex, which typically functions as a toxin-antitoxin system. Recent studies reveal a consistent defense mechanism: RT-msDNA complex functions as a molecular sentinel that senses phage-encoded triggers and regulates effector activation. Perturbation of RT-msDNA unleashes diverse cytotoxic effectors, thus inducing abortive infection (Abi) to restrict viral propagation. This review summarizes recent advances in retron activation mechanisms, effector diversity, and structural assembly, providing a comprehensive perspective on retron-mediated immunity.
|
|
Scooped by
mhryu@live.com
July 18, 10:31 AM
|
Fermented foods exemplify self-organized microbial ecosystems shaped by ecological filtering, domestication, and sustained human selection. This review explores how ecological insights from traditional fermentations guide the rational design of engineered microbial consortia in contemporary food biotechnology. Across various substrates and cultural contexts, fermented systems illustrate that functional stability, metabolic complementarity, and reproducible biochemical outputs arise from structured community interactions rather than from individual strains. Recent advances in multiomics, culturomics, and genome-scale metabolic modeling facilitate the systematic identification of core microbiota and ecosystem-level functional phenotypes that underpin fermentation performance. These developments are shifting fermentation from an empirical practice to a predictive, design-oriented discipline. Integrating ecological theory with systems-level biotechnology, traditional fermentations offer conceptual and experimental frameworks for constructing minimal yet robust microbial consortia. Such strategies enable the development of next-generation fermented foods with enhanced nutritional, functional, and sensory attributes, while preserving ecological robustness and process reproducibility.
|
|
Scooped by
mhryu@live.com
July 17, 3:33 PM
|
Invasive aspergillosis most commonly manifests as pulmonary disease. However, in a subset of patients, the fungus disseminates from the lungs to secondary sites of infection. Disseminated disease is associated with markedly worse clinical outcomes than pulmonary infections, underscoring the importance of understanding the biological mechanisms through which Aspergillus can escape from the pulmonary environment. Here, we review the molecular and cellular events that facilitate dissemination of Aspergillus fumigatus, with a focus on interactions at the host–pathogen interface within the vasculature. Additionally, we examine how fungal morphogenesis, adhesion to endothelial surfaces, and penetration of vascular barriers contribute to hematogenous spread. Finally, we highlight areas of open investigation to focus future research efforts.
|
|
|
Scooped by
mhryu@live.com
July 18, 11:47 PM
|
Antimicrobial resistance (AMR) is increasingly recognized as a One Health challenge driven by the continuous exchange of resistant bacteria and resistance determinants across human, animal, and environmental sectors. While genomic surveillance has substantially improved detection of antimicrobial resistance genes (ARGs), most monitoring frameworks remain gene- or isolate-centric, limiting insight into the mechanisms that govern resistance transmission and persistence. Recent evidence indicates that plasmids, self-replicating mobile genetic elements (MGEs) capable of horizontal transfer across bacterial species, play an important role in disseminating clinically relevant resistance determinants across sectors. In this mini-review, we synthesize genomic and ecological evidence demonstrating that a limited number of plasmid incompatibility (Inc) groups recur across human, animal, and environmental reservoirs, often independent of bacterial host lineages. We highlight how plasmid transmission dynamics are shaped by host-independent mobility, ecological generalism, co-selection with accessory traits, and persistence in engineered and natural environments. We further examine why current AMR surveillance approaches, including ARG-centric metagenomics and isolate-based monitoring, systematically overlook these plasmid-mediated processes. Furthermore, we propose that plasmid-resolved analysis represents a critical and currently underutilized complementary layer for One Health AMR surveillance. Integrating plasmid classification and genomic reconstruction into wastewater-based epidemiology and cross-sector monitoring frameworks can improve attribution of transmission pathways, enhance early detection of high-risk resistance, and provide a mechanistic foundation for risk-informed intervention strategies.
|
|
Scooped by
mhryu@live.com
July 18, 11:41 PM
|
A vast portion of genes in microbial genomes, termed the “unknome,” remains functionally uncharacterised. This genetic “dark matter” represents a significant bottleneck in microbiology, as it is often excluded from genomic studies. We argue that a substantial part of the unknome encodes functions critical for biotic interactions, the complex dialogues among microbes or between microbes and their hosts. These functions are rarely observed under standard laboratory conditions, which rely on simplified pure cultures. Unlocking the unknome therefore calls for a stronger emphasis on ecologically relevant experimental systems. By embracing complexity through co-culture and in situ analyses, we can begin to decipher this hidden genetic repertoire, deepening our understanding of microbial communication, adaptation, and evolution. Crucially, the conceptual and methodological challenges raised by the microbial unknome resonate well beyond microbiology: parallel “dark” fractions of uncharacterized genes and proteins pervade eukaryotic genomes, from lineage-specific (orphan) genes underpinning novelties in plants and animals, including humans. Embracing ecological and systems-level approaches to dissect the unknome therefore has the potential to reframe how we link genotype to phenotype in context-dependent, interaction-driven biological systems.
|
|
Scooped by
mhryu@live.com
July 18, 4:56 PM
|
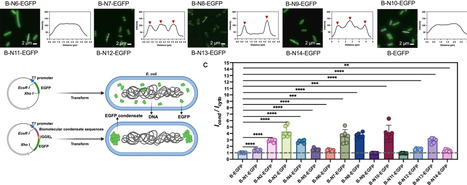
Biomolecular condensates form through phase separation, playing a crucial role in cellular organization and gene regulation. However, native condensate elements often suffer from high molecular weight and sequence redundancy, limiting their use in prokaryotic systems. To address this, we established an integrated framework combining generative artificial intelligence, computational prediction, and experimental validation. We began by constructing a benchmark dataset of eukaryotic-derived phase-separation elements in E. coli. This dataset was used to build the PSVAE model for de novo sequence design and the LMPsPred classifier for high-throughput screening. Finally, we identified 13 short-sequence elements with low molecular weight (16-26 kDa) and minimal redundancy. Experimental validation confirmed that these elements formed condensates in E. coli, with enhanced protein recruitment compared to native elements. This study highlights the potential of AI-guided design to generate condensate-like assemblies and expands the repertoire of candidate phase-separation elements for prokaryotic systems. llps
|
|
Scooped by
mhryu@live.com
July 18, 4:49 PM
|
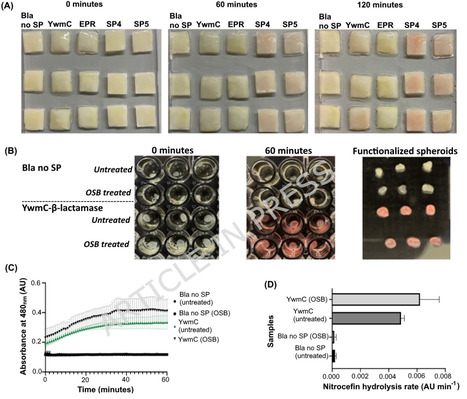
Bacterial nanocellulose (BC), produced by Komagataeibacter species, is an ideal scaffold for biological Engineered Living Materials (ELM) research. Current BC functionalization strategies often rely on secondary microbial hosts or post-production enzyme immobilization, limiting the scalability and modularity required for programmable bioELMs. Establishing a single-chassis system capable of simultaneous biopolymer synthesis and in situ functionalization remains a primary objective in bioELM research. This study addresses the need by benchmarking signal peptide-mediated protein translocation in K. rhaeticus iGEM, a model bacterium for BC-based bioELMs, enabling a synthetic biology framework for single-chassis based biomaterial functionalization. Genome-wide analysis confirmed the presence of a complete Sec translocation machinery in K. rhaeticus. Through liquid chromatography-tandem mass spectrometry and SignalP 5.0 prediction, native signal peptides were identified and evaluated alongside previously characterized heterologous signal peptides using β-lactamase and mScarlet as cargo proteins. Protein translocation was found to depend on signal peptide identity, cargo type, and expression mode. Fluorescence imaging revealed cytoplasmic, polar, and peripheral localization patterns, confirming functional engagement with the native translocation machinery. A key limitation identified was the retention of recombinant proteins within the periplasm, restricting extracellular availability. Despite this, signal peptide-mediated translocation enabled the incorporation of enzymatic activity into BC during biosynthesis. A post-growth osmotic shock-release strategy increased measurable enzymatic activity by 30%, demonstrating a practical route to overcome this physiological bottleneck while maintaining the biomaterial production capacity. This study benchmarks signal peptide-dependent protein translocation in K. rhaeticus and identifies periplasmic retention as a key constraint for extracellular protein release. By linking protein translocation to in situ BC functionalization, this work establishes a synthetic biology framework that supports the development of K. rhaeticus as a single-chassis platform towards the production of functionalized bioELMs.
|
|
Scooped by
mhryu@live.com
July 18, 4:34 PM
|
Large language models (LLMs) for biological sequences are transforming computational biology, enabling a nuanced understanding of protein and nucleotide sequence data. Recent models, including ESM2, ESM3, AlphaGenome, Evo-1, and Evo-2, adapt natural language processing principles to the biological domain by learning high-dimensional hidden representations that capture evolutionary constraints, structural patterns, and functional motifs. This mini-review summarizes recent developments in devising and applying such models, emphasizing viral protein analysis. We highlight studies that have leveraged sequence-based LLMs in the protein domain (i.e. protein language models, or PLMs) for important application tasks such as viral protein annotation, variant effect prediction, and immune escape characterization. Additionally, we present a benchmark evaluation of these state-of-the-art protein language models to evaluate their core ability to capture evolutionary relationships between viral protein sequences. By discussing the opportunities and challenges of PLMs, the review outlines a road map for the potential application of LLMs in empowering virology research and pathogen surveillance.
|
|
Scooped by
mhryu@live.com
July 18, 4:24 PM
|
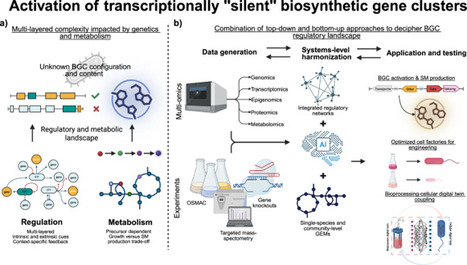
Microbial biosynthetic gene clusters (BGCs) encode diverse bioactive molecules but remain mostly silent under lab conditions, reflecting a balance between regulatory control and the metabolic cost of secondary metabolite production. While activation strategies exist, the yields stay low because switching on a cluster doesn’t guarantee the host can sustain biosynthesis. This review covers the regulatory architecture and metabolic constraints, including precursor availability, energy status, and nutrient sensing, that shape BGC output. We argue that coupling regulatory network models with genome-scale metabolic models offers a powerful framework for unlocking silent BGCs and realising the full biosynthetic potential within microbial genomes. This review summarises multilayered regulatory and metabolic systems governing silent biosynthetic gene clusters in microbes, and how coupling regulatory network models with genome-scale metabolic models can unlock their biosynthetic potential
|
|
Scooped by
mhryu@live.com
July 18, 4:15 PM
|
The Klebsiella oxytoca Species Complex (SC) represents an emerging healthcare-associated group of opportunistic pathogens. The capsular polysaccharide is a virulence determinant and target for novel vaccines, monoclonal antibodies and phage therapy. In the absence of broadly accessible phenotyping techniques, prediction of capsule types from whole-genome sequence data is critical for understanding capsule diversity and epidemiology, and to prioritise capsule types as targets for novel anti-K. oxytoca SC interventions. Here we present the first comprehensive capsule synthesis locus (K locus) database targeted for the K. oxytoca SC, comprising 88 distinct loci defined by gene content and which is compatible with the rapid genome typing tool Kaptive. The database provides high coverage of publicly available K. oxytoca SC genomes (97.6% of 2,244 genomes, dereplicated from a total of 4,055), and the typing rate is significantly higher than that achieved with the pre-existing Klebsiella K locus database (97.6% vs 50.3%, p <0.0001), which primarily targets the Klebsiella pneumoniae SC. We demonstrate the utility of the novel K. oxytoca SC database by application to three diverse clinical K. oxytoca SC isolate collections (n=61 to 102 genomes each), suggesting a high diversity of K types. The novel K. oxytoca SC K locus database (github.com/klebgenomics/KoSC-surface-antigen-loci) will provide a key resource to support larger systematic studies and ongoing genomics surveillance efforts for the K. oxytoca SC.
|
|
Scooped by
mhryu@live.com
July 18, 12:43 PM
|
High molecular weight fibrous proteins such as silk, elastin, and collagens, are fundamental for providing shape to macroscopic biological structures, yet their recombinant production remains challenging because of their extreme size and sequence repetitiveness. Here, we report a circular RNA-based ribosome translation platform that enables iterative ribosome synthesis of fibrous proteins through continuously 'looped' translation. To promote efficient circularization of repetitive fibrous protein-transcripts, we combined a synonymous codon locker sequence strategy with RNA circularization chaperones. Guided by a ribosome traffic model, we further optimized the translation bottlenecks within the circular RNA, substantially improving translation yields. The established looped translation platform is applicable to at least six classes of fibrous proteins and generated products with molecular weight exceeding titin at 3.8 MDa. The synthesized polypeptides were characterized through electron microscopy, bulk material fabrication, and mechanical analysis, demonstrating properties associated with ultra-high molecular weight polypeptides. Finally, we coupled looped translation to secretion through a programmed ribosomal frameshift, enabling export of fibrous protein across cellular membranes in both Escherichia coli and Bacillus subtilis. We envision that the genetic tools presented here could find a range of applications in bioplastics and engineered living materials.
|
|
Scooped by
mhryu@live.com
July 18, 10:56 AM
|
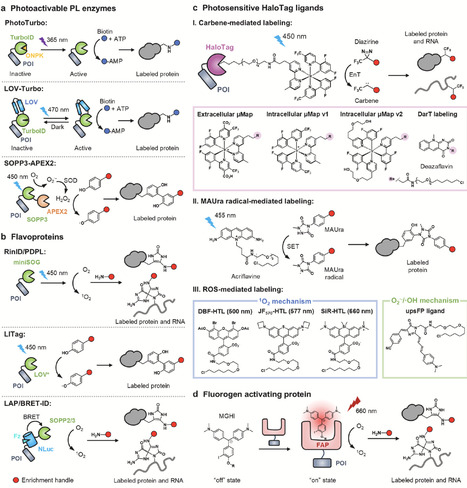
Proximity labeling (PL) has become a broadly used chemical strategy for mapping molecular neighborhoods in living systems. Recent advances are shifting the field from expanding the PL toolbox toward engineering the reaction boundary that defines what is labeled, when labeling occurs, and how far reactive intermediates propagate. In this review, we highlight recent developments in four areas. First, new genetically encoded enzymatic systems reduce reliance on classical peroxide- or biotin-dependent chemistry. Second, optically controlled PL methods improve temporal gating through photoactivated enzymes, genetically encoded photocatalysts, self-labeling tag ligands and fluorogen-activating protein systems. Third, molecular ruler strategies and engineered enzyme-probe pairs are refining our understanding and control of labeling radius. Finally, emerging platforms repurpose covalent labeling for signal integration and functional amplification. Together, these advances position PL as a programmable chemical tool for spatial mapping, molecular recording and biological intervention.
|
|
Scooped by
mhryu@live.com
July 18, 10:47 AM
|
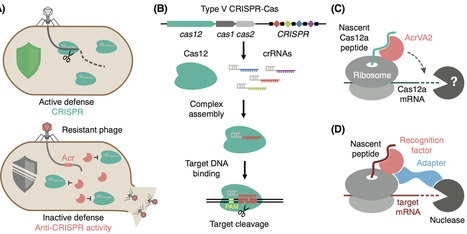
Mobile genetic elements and their hosts engage in continuous evolutionary conflict. Marino et al. recently uncovered an unusual anti-CRISPR mechanism: the phage protein AcrVA2 triggers translation-coupled mRNA degradation by recognizing nascent Cas12. The findings suggest that nascent peptides may signal an underappreciated layer of gene regulation across the kingdoms of life.
|
|
Scooped by
mhryu@live.com
July 18, 10:36 AM
|
Fermented foods are produced from established bioprocesses where microbes convert raw materials into safe, sensory-rich products. Multi-omics can help elucidate how fermentation dynamics shape metabolites and microbial components that have the potential to influence the gut microbiota and host metabolism. The practical bottleneck is translation: many omics observations remain associative, and translating them into health-directed starter cultures requires verification of product-level markers, demonstration of process scalability, and compliance with regulatory requirements. Health-directed starter cultures are defined as single strains or designed consortia selected to control fermentation while enriching a small set of trait axes, such as indole-derivative formation and bile-acid transformation. Here, we propose a stepwise framework that connects genome-encoded functional capacity and genomic safety assessment with pathway execution in the target matrix, quantitative product chemistry, and mechanism-aligned functional assays. Lastly, we outline requirements for human trials, emphasizing individual variability and the need for study designs that connect quantified food components to measurable health benefits.
|
|
Scooped by
mhryu@live.com
July 18, 10:23 AM
|
We survey recent works on designing and implementing CO2 fixation pathways. We describe how these studies reveal a gap between a well-designed pathway and a productive strain. This gap includes phenomena that are often not accounted for in design, such as hidden enzyme energy usage and regulatory rewiring during adaptive evolution. Several systems biology tools can help overcome this gap. Kinetic models can be generated rapidly using new algorithms, and when coupled with retrobiosynthesis, can evaluate pathway dynamics and stability in the context of the native network. Quantitative and interaction proteomics reveal how the host strain adapts to novel pathways and can identify interference from native metabolites. In vivo optimization through adaptive laboratory evolution is used extensively to optimize modules or whole pathways, but requires the pathway phenotype to be linked to growth. Post-evolution characterization reveals that cells optimize pathway integration through substantial proteome reallocation.
|


















mcf