 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:11 AM
|
Assembly-line polyketide synthases (PKSs) are among the most sophisticated catalysts in nature, responsible for the biosynthesis of many medicinally important natural products including antibiotics, anticancer agents, immunosuppressants and veterinary agents. While conventional methods for polyketide natural product discovery have fallen out of favor, the rapid expansion of microbial genome sequencing has revealed a vast untapped diversity of assembly-line PKSs, most of which can be regarded as “orphans” in that their product identity is unknown. A clear picture of how this sequence diversity reflects the evolutionary history of assembly-line PKSs could enable judicious prioritization of experimental efforts aimed at decoding these orphans. To this end we have used a scalable curation workflow to hand-curate an updated catalogue, PKSClusterDB, that includes 16 633 non-redundant assembly-line PKSs. We have also formulated an automatable “anchor-window” framework based on conserved multimodular segments of assembly-line PKSs to identify and interrogate PKS families of interest. Application of this framework to three different PKS families extracted from PKSClusterDB revealed lineage-dependent patterns of diversification, ranging from broadly diversified families to more compact or sharply bounded lineages. PKSClusterDB has also proven useful in exploring other aspects of PKS diversity, including scaffold architecture, extender-unit specificity, reductive-state programming, stereochemical control and modular organization. In closing, we consider how advances in machine learning could be harnessed to accelerate our understanding of assembly-line PKS evolution, diversity and biosynthetic mechanisms.
|
|
Scooped by
mhryu@live.com
June 27, 11:55 PM
|
Antibiotics are among medicine’s greatest successes, but resistance evolution threatens their continued efficacy. Decades of research have deepened our understanding of the mechanisms and evolutionary dynamics of antimicrobial resistance. More recently, advances in machine learning (ML) and artificial intelligence (AI) show promise in predicting antimicrobial resistance in pathogens based on rapid whole-genome sequencing and other accessible data. In this perspective, we highlight advances in understanding the mechanisms and spread of antimicrobial resistance. We discuss how this knowledge, coupled with ML- and AI-based approaches, can inform the prediction of resistance and a precision-medicine strategy that targets pathogenic bacteria specifically, thereby limiting resistance evolution and collateral damage to the microbiome. These accurate predictions of bacterial vulnerabilities will enable the adaptation of classical antimicrobial treatments with adjuvants, as well as the use of novel, narrow-spectrum therapeutics. Implementing these strategies, while also identifying key challenges, will help bring this strategy into clinical practice.
|
|
Scooped by
mhryu@live.com
June 27, 11:50 PM
|
Trichoderma is a genus of beneficial fungi widely used in agriculture disease suppression, plant growth promotion, and soil health improvement. Although some studies have shown that Trichoderma spp. can be seed-borne, albeit at low frequency, no specific study has so far focused on the diversity of Trichoderma isolates capable of adapting to this particular environment. Over 2 years of routine seed health analyses, we isolated 107 Trichoderma strains associated with seeds from 32 cultivated plant species, and we investigated the diversity of this specific library using multi-locus analysis. These isolates are distributed among 18 different species, but Trichoderma atroviride is by far the most frequent. Representative strains of each species all have the ability to parasitise damping-off agents, although the intensity of parasitism can vary greatly from one strain to another and depending on the targeted plant pathogens. For two isolates of T. atroviride, we showed that these fungi can be transmitted from fruit to seed and limit the transmission of the plant pathogen Alternaria brassicicola. Furthermore, these strains stimulated seedling growth and provided protection against Globisporangium ultimum. Together, these findings support the benefits of focusing on certain selected seed-borne Trichoderma which seem particularly well suited to improving seed health.
|
|
Scooped by
mhryu@live.com
June 27, 11:38 PM
|
Microbial community assembly is shaped by the nature of available resources, with labile carbon sources like glucose often expected to support low diversity due to rapid growth and competitive exclusion. In contrast, recalcitrant substrates like cellulose are thought to support higher diversity through slower growth and increased niche partitioning. In previous work, we showed that compost-derived microbial communities propagated on cellulose maintained high diversity over nearly a year. To determine whether such diversity is specific to recalcitrant substrates or reflects more general features of assembly, we tracked community dynamics in three environments—cellulose paper, cellulose broth and glucose—using daily 16S rRNA profiling. Communities maintained through four bi-weekly serial transfers, with five replicates per treatment, yielded a high-resolution dataset of over 800 samples. Despite originating from the same inoculum, communities diverged sharply in both taxonomic and functional composition. Cellulose environments yielded stable communities enriched in specialists, while glucose environments exhibited rapid succession and dominance by generalists. Surprisingly, all environments sustained comparably high levels of taxonomic diversity. Functional inference suggested extensive cross-feeding and resource salvaging in both cases. Our results reveal distinct assembly trajectories under simple carbon regimes and provide a foundation for future mechanistic study.
|
|
Scooped by
mhryu@live.com
June 27, 11:28 PM
|
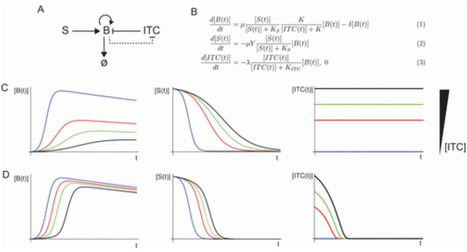
Healthy plant leaves potentially host both commensal bacteria and opportunistic pathogens, which, under some circumstances, may cause disease. The interactions between commensals and opportunistic pathogens are generally poorly understood, but such understanding is crucial for developing effective biocontrol strategies. In Arabidopsis thaliana, isothiocyanates (ITCs) are defense metabolites that suppress most bacteria; commensals are especially affected as they do not express ITC resistance genes. The ITC hydrolase SaxA detoxifies ITCs, making it an important virulence factor for bacterial and fungal pathogens. To investigate pathogen-commensal interactions based on SaxA-mediated ITC degradation, we used five ITC-sensitive bacterial commensals and the opportunistic pathogen Pseudomonas viridiflava 3D9 (PS). All strains were isolated from healthy A. thaliana leaves. PS degrades 4-methylsulfinylbutyl-ITC (4MSOB-ITC) with SaxA. We examined commensal growth in the presence of 4MSOB-ITC, both in monoculture and in coculture with PS or a saxA-deficient mutant (PSKO). We used the growth data to develop a generalizable consumer-resource mathematical model incorporating ITC toxicity, ITC degradation, and nutrient use. We predicted and confirmed experimentally that the extent to which SaxA benefits the pathogen depends on its effects on commensals. In some contexts, commensal rescue and the resultant nutrient competition limit pathogen growth. In addition, we tested in silico how commensal ITC susceptibility, pathogen ITC degradation rates, and growth parameters affect the trade-off between SaxA-mediated virulence (strong pathogen growth) and commensal rescue (commensal growth). Our findings suggest that the effects of microbial traits—traditionally viewed as either virulence or plant-beneficial factors—are constrained in the microbiome context. This underscores the need to reconsider how such traits are classified in the context of plant-microbiome interactions.
|
|
Scooped by
mhryu@live.com
June 27, 4:28 PM
|
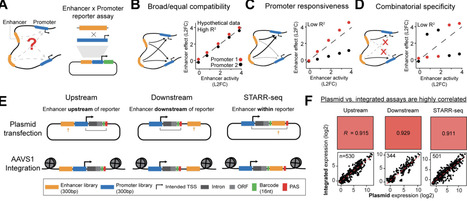
Enhancers activate specific target promoters, but whether intrinsic enhancer-promoter compatibility contributes to this specificity is debated. Recent studies using different reporter assays have reached contradictory conclusions. We compare six reporter assay designs, identify confounders that bias compatibility measurements, and apply improved assays to test 25,000 enhancer-promoter pairs. Promoters differ in their responsiveness to enhancers (>100-fold versus 1.1-fold activation) while enhancers activate all promoters in a similar rank order. Promoter output scales with enhancer activity following a power law, with the exponent varying across promoters. Incorporating this exponent into the Activity-by-Contact model improves prediction of endogenous enhancer effects, explaining why certain active genes are insensitive to distal perturbations and "skipped" by enhancers. Responsiveness is modulated by transcription factor motifs in the core promoter. This work establishes responsiveness as an intrinsic promoter property that enables specific promoters to be highly activated in a landscape of broadly compatible enhancers, while others remain unaffected.
|
|
Scooped by
mhryu@live.com
June 27, 4:17 PM
|
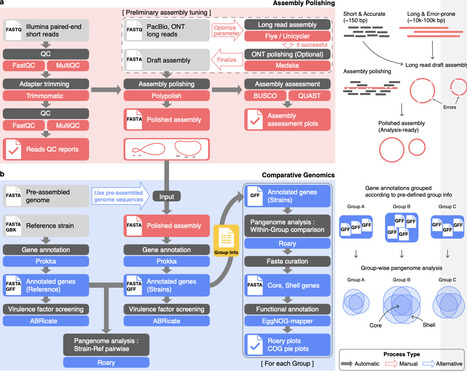
Organisms have continuously evolved in response to environmental conditions. Pathogenic bacteria evolve under host and environmental pressures, reshaping their genomes through insertions, inversions, deletions, and duplications during infection. In clinical settings, phenotypic traits of pathogenic bacteria such as virulence or antimicrobial resistance directly affect disease severity, transmission, and treatment. Conventional genotyping provides insights into genomic relatedness but does not always align with these clinically relevant traits, limiting its utility for phenotype-driven interventions. Here, we develop ABComp (Assembly polishing and Bacterial whole-genome Comparison for multi-group clinical isolates), a modular and Snakemake-based workflow for phenotype-driven comparative genomics. ABComp automates assembly polishing, group-wise pangenome analysis, and enables flexible pathogenic marker discovery through user-defined comparisons. We validated ABComp using a Klebsiella pneumoniae ground truth dataset stratified by yersiniabactin presence and successfully recovered the entire locus as a group-specific core marker. By applying ABComp to another dataset of clinical isolates with experimentally measured virulence, we discovered the ferric citrate (Fec) uptake system as a potential marker specific to a hypervirulent group. These results demonstrate ABComp's utility in uncovering phenotype-linked genomic markers with clinical significance, supporting targeted treatments and rapid diagnosis.
|
|
Scooped by
mhryu@live.com
June 27, 4:06 PM
|
High-quality plant genome assemblies are rapidly increasing, but accurate structural annotation remains reliant on transcript and homology evidence, limiting applications in newly sequenced and non-model species. Here, we present PlantGeneAnn, a plant-optimized, strand-specific genome foundation model for ab initio gene structure annotation. Fine-tuned on only nine high-quality model plant annotations, PlantGeneAnn outperformed a multi-species model trained on 42 species, showing that annotation quality is more important than token volume. On a stringent 13-species benchmark covering rosids, asterids, and monocots, PlantGeneAnn surpassed four state-of-the-art baselines across five evaluation levels, from base-level classification to complete transcript recovery. It achieved higher intron precision and better captured complex gene structures. In zero-shot variant effect prediction, PlantGeneAnn identified cryptic splice donors and premature stop codons in maize and rice, with saturation mutagenesis confirming single-nucleotide, context-dependent sensitivity. It also retained generalizability for epigenomic track prediction, highlighting its value for pan-genomics, crop improvement, and non-model plant research.
|
|
Scooped by
mhryu@live.com
June 27, 3:27 PM
|
Protein self-assembly is a fundamental biological process of great importance for the design and synthesis of biomaterials. Developing the ability to precisely manipulate protein assembly would greatly expand both our understanding of the process and our biotechnological capabilities. Within bacteria, proteins that self-organize to form bacterial microcompartments (MCPs) offer an excellent model system for studying protein self-assembly and advancing biomaterial design capabilities. MCPs consist of irregular polyhedral shells that encase an enzyme core that acts as enzymatic nanoreactors. In isolation, the abundant shell proteins of the 1,2-propanediol utilization (Pdu) MCP, PduA and PduJ, have a high propensity to self-assemble into tubular structures, analogous in form to carbon nanotubes. Here, we modulate higher-order assembly of PduA and PduJ hexamers by systematically altering their charge through charge inversion and supercharging across multiple platforms including heterologous overexpression and cell-free protein synthesis. Overexpression and cell-free experiments show that increasing the overall negative charge of assembling subunits consistently promotes self-assembly into tubular structures. Using molecular simulations, we determined the preferred bending angle adopted by the hexameric proteins to predict the most probable self-assembled structures, including honeycomb-like sheets and nanotubes. Simulations of closed PduA and PduJ tubes show the interactions responsible for tube stability, chirality, and radius. In vivo, we find that these charge-altered hexamers are assembly competent within the native MCPs in Salmonella enterica LT2. Our results collectively reveal that both electrostatic interactions and fields generated by charges on proteins can be leveraged to control protein-based nanostructures.
|
|
Scooped by
mhryu@live.com
June 27, 3:04 PM
|
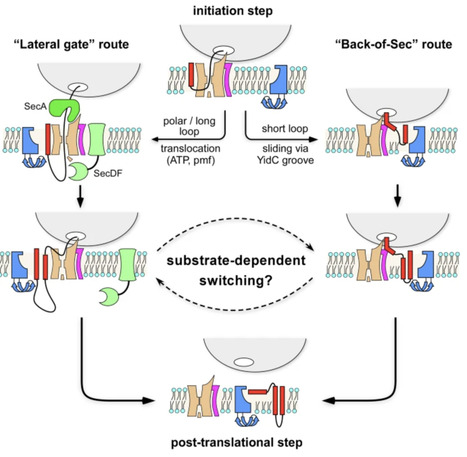
The Sec translocon and the YidC/Oxa1-type insertases universally mediate biogenesis of α-helical membrane proteins, but the molecular basis of their cooperation has remained disputed. Recent discovery of multi-subunit insertases assembled at the back of the translocon in fungi and higher eukaryotes has raised questions about the architecture and mechanism of the putative bacterial ortholog SecYEG-YidC. Here, we combine cryogenic electron microscopy with cell-free protein synthesis to visualize biogenesis of the SecYEG/YidC-dependent multipass membrane protein NuoK. The nascent chain of NuoK does not enter the lateral gate of SecYEG but instead crosses the translocon towards its back side, where YidC is recruited in the nascent substrate-dependent manner. The SecY-YidC interface promotes folding of the transmembrane helices before insertion, consistent with thermodynamic principles of membrane protein folding. YidC forms extensive contacts with the nascent chain, suggesting its key role in the insertion event. These findings provide mechanistic insight into membrane protein insertases, support evolutionary conservation of a gate-independent insertion route, and expand current models of membrane protein biogenesis.
|
|
Scooped by
mhryu@live.com
June 27, 2:39 PM
|
Type II topoisomerases are essential enzymes that alter DNA topology by making a transient double-stranded break in one segment of the DNA and passing another DNA segment through the break such that the sign of the crossing DNA segments is inverted. DNA gyrase has served as a paradigm for mechanistic studies of topoisomerase action. Prescient biochemistry predicting a positive sign for the crossing DNA segments during DNA gyrase supercoiling activity has recently been confirmed by cryo-electron microscope studies of the enzyme bound to DNA. These studies demonstrated a central role of the C-terminal domain of the GyrA subunit in shaping the DNA crossover. We show biochemically that exchanging the DNA GyrA CTD for the homologous one from the ParC subunit of Topoisomerase IV alters the orientation of the DNA crossover from a positive one to a negative one. The sign of the pre-catalytic DNA crossover in Topoisomerase IV is also negative. We suggest that relaxation of negatively supercoiled DNA by Topoisomerase IV proceeds by the opposite of the mechanism by which DNA gyrase supercoils DNA.
|
|
Scooped by
mhryu@live.com
June 26, 4:01 PM
|
Temperate bacteriophages are dominant members of the human gut microbiome that can infect and lyse their bacterial hosts or integrate as prophages. During this integrated state, prophages exhibit extensive control over host physiology and lysis via induction. Here, we studied a diverse collection of Bacteroidales isolates, which are amongst the most abundant bacterial orders within the human gut, identifying 902 high-quality prophage genomes present within 305 isolates, 240 of which were poly-lysogens. Despite their prevalence, our understanding of the function and induction triggers of prophages is limited. To predict prophage induction, we employed an iterative profile Hidden Markov Model search across divergent bacterial hosts to identify prophage regulatory components. We found 197 Bacteroidales prophages encoding complete CI-like repressor proteins, which initiate induction upon DNA damage. We selected Bacteroides thetaiotaomicron strain Bt_806 to characterise further as it harbored six diverse prophages, including the prevalent and abundant prophage LoVE, which was the only integrated prophage encoding a complete CI-like repressor. Transcriptomics revealed phage LoVE was routinely induced upon DNA damage, while the five co-habiting prophages remained stably integrated yet exhibited transcriptionally active genes associated with regulation, prophage maintenance, and uncharacterised functions. Finally, we selected an additional eleven Bacteroidales poly-lysogens, confirming that integrated prophages encoding complete CI-like repressors were reliably induced upon DNA damage. Together, we demonstrate that mechanistic understanding of prophage induction linked with identification of regulatory genes enables selective and predictable induction of gut prophage species as a potential tool to modulate the microbiome.
|
|
Scooped by
mhryu@live.com
June 26, 3:42 PM
|
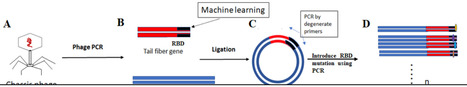
Klebsiella pneumoniae (Kp) is a leading cause of bacterial nosocomial infections. The rapid emergence of multidrug-resistant (MDR) Kp strains poses a significant global health threat, challenging current antibiotic-based therapies. While bacteriophage therapy offers a promising alternative, its effectiveness is often limited by the narrow host ranges of natural phages and the rapid emergence of phage resistance. Synthetic phages, with their diverse and customizable genomes, have the potential to overcome these limitations. In this study, we engineered synthetic bacteriophage variants using two in-house phage isolates, ɸ115 and ɸ100, which exhibited limited efficacy against a range of clinical Kp strains. In contrast, the synthetic phage libraries derived from the recombination of tail fiber genes and genomes of ɸ115 and ɸ100 exhibited a significantly extended host range and the ability to lyse phage resistant Kp strains. Indirect measurement of bacterial respiration as a growth indicator in presence of phage, using the OmniLog system revealed that no phage resistance Kp emerged against the synthetic phage libraries within 24 h, while other Kp strains developed resistance to the parental phages. Host range analysis, burst size measurements, and genomic DNA comparisons of individual synthetic phage isolates indicated that a short central tail fiber of ɸ100 likely plays a pivotal role in extending the host range and lysing resistant Kp strains. Our findings highlight the potential of synthetic bacteriophages to overcome the dual challenges of narrow host specificity and phage resistance. This represents a significant advancement toward developing viable phage therapeutics against MDR Kp infections.
|
|
|
Scooped by
mhryu@live.com
Today, 12:01 AM
|
Chimeric natural products are formed when biosynthetic inputs from distinct pathways, gene clusters, or metabolic branches are integrated into a single scaffold. In bacteria, such crosstalk-driven assembly can generate structurally diverse metabolites with distinct biological activities. However, these metabolites remain underexplored because most discovery pipelines are optimized to connect one metabolite family to one co-localized biosynthetic gene cluster (BGC). As a result, metabolites produced through inter-pathway collaboration, recruitment of primary-metabolic intermediates, or non-enzymatic coupling of products from separate pathways are often deprioritized as genome-metabolome mismatches. Here, we describe chimeric natural products as an important yet overlooked class of bacterial metabolites and present a discovery framework that prioritizes candidate hybrid scaffolds from LC-MS-based metabolomics. We suggest that public-repository spectral searching, multi-class MS/MS annotation, retrospective genome-to-metabolite linkage, and isotope-guided validation can improve discovery of these overlooked scaffolds. Prioritizing such metabolites can expand natural product discovery beyond canonical frameworks, uncover new biosynthetic design principles, and inform future efforts to engineer hybrid molecules.
|
|
Scooped by
mhryu@live.com
June 27, 11:53 PM
|
Pseudomonas aeruginosa is an opportunistic pathogen responsible for acute and chronic infections and is characterized by a remarkable ability to develop antibiotic resistance. This has prompted increasing interest in alternative or complementary antimicrobial strategies, including the use of essential oils (EOs), which are complex mixtures of plant-derived volatile compounds with documented biological activities. This review aims to summarize and discuss the current evidence on the antimicrobial, antibiofilm, and anti-virulence activities of EOs against P. aeruginosa, with particular emphasis on their mechanisms of action, synergistic interactions with antibiotics, and limitation to the possible clinical applicability. A large number of studies, mainly conducted in vitro, indicate that EOs and their major constituents can impair P. aeruginosa viability and pathogenicity through multiple mechanisms. In addition, several EOs or purified terpene components exhibit synergistic effects when combined with conventional antibiotics, enhancing antimicrobial efficacy. However, in vivo evidence remains limited, largely restricted to topical infection models, while clinical studies in humans are currently lacking. Significant challenges related to chemical variability, toxicity, safety, standardization, and regulatory classification also emerge from the literature. Although EOs show considerable promise as antimicrobial and anti-virulence agents against P. aeruginosa, their current role should be viewed primarily as adjunctive or alternative strategies rather than as standalone systemic therapies. Future progress will depend on the development of standardized and safer formulations, advanced delivery systems, and well-designed in vivo and clinical studies.
|
|
Scooped by
mhryu@live.com
June 27, 11:40 PM
|
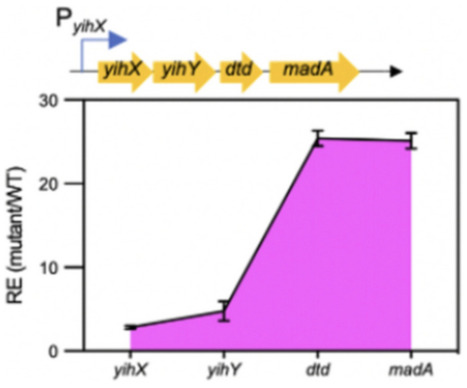
The E. coli fabH gene encodes the 3-ketoacyl-ACP synthase III that initiates fatty acid synthesis. Deletion of the fabH gene results in severely limited fatty acid synthesis and tiny cells that are unusually sensitive to antibiotics. Genetic investigations identified the yiiD gene, now called madA, that encodes a malonyl-ACP decarboxylase that suppresses the ∆fabH phenotype, but only when madA is multicopy. We selected vancomycin-resistant suppressor derivatives of a ∆fabH strain. Although such chromosomal mutations were generally rare and weak, suppressor strain s1 (a single C-T transition) restored both wild-type growth, high-level vancomycin resistance, and wild-type fatty acid synthesis by creation of a new promoter within the coding sequence of a gene upstream of madA. This provides a caveat to the extensive effort to develop FabH inhibitors as antibacterial drugs.
|
|
Scooped by
mhryu@live.com
June 27, 11:31 PM
|
Cell-to-cell communication in microbial systems is known for its vital role in cellular signalling and gene expression. A specific form termed Quorum Sensing (QS) has received considerable attention since its discovery in the marine symbiont Aliivibrio fischeri. QS-controlled microbial functions are associated with bacterial virulence, pathogenicity, host–microbe interactions, and biofilm development. Interference in these signaling systems can modulate microbial virulence and pathogenicity, and microbial infection caused by drug-resistant pathogens. Plant-derived phytochemicals are considered a promising candidate, with coumarins emerging as significant plant-derived signalling molecules shaping microbiome dynamics and pathogen behaviors from a broad spectrum of ecosystems. Here we explored the role of natural and synthetic coumarin compounds in the control of signalling and virulence traits in Pseudomonas aeruginosa and other priority bacterial pathogens, including the fungal opportunist Aspergillus fumigatus. We uncovered an important ‘hydroxylation-bias’ favoring coumarin, umbelliferone (7-OH), and 6-hydroxy-coumarin (6-OH) in the specific competitive inhibition of the Pseudomonas Quinolone Signal (PQS), associated with reduced activity of a PqsR translational fusion and suppression of pyocyanin production. Conversely, while esculetin (6,7-OH) was most effective at Acyl Homoserine Lactone (AHL) QS biosensor inhibition, it did not affect PQS production. Anti-biofilm activity of coumarins against P. aeruginosa was independent of initial attachment but linked to changes in exopolysaccharide production. As the very real threat posed by antimicrobial resistance persists, these data support a role for phytochemicals such as coumarins in delivering an ecological solution to dysbiosis in the host–microbe interaction.
|
|
Scooped by
mhryu@live.com
June 27, 5:25 PM
|
Understanding genome regulation is limited by the complexity of molecular interactions in living cells. Cell-free systems provide a simplified platform for studying gene expression, but low mRNA levels have prevented RNA-seq. To address this, we develop an active learning workflow combining Bayesian optimization with automated high-throughput experimentation to systematically explore over 1.6 million buffer compositions, experimentally testing 653. We identify a “mRNA-optimized” buffer (20-fold increase in mRNA yield) and a “trade-off” buffer (13-fold increase while maintaining protein production). Using direct RNA-seq, we profile the T7 phage transcriptome in cell-free systems and compare it with a purified T7-RNAP transcription system and phage-infected bacteria. This comparative analysis reveals distinct regulatory layers: the T7-RNAP system captures promoter-strength hierarchies but lacks RNA degradation, whereas cell-free systems provide an accurate estimation of in vivo expression and reveal mRNA maturation sites. This work establishes cell-free transcriptomics as a controlled approach to study genome regulation. Cell-free is a powerful platform to unravel biological complexity. Here, the authors used active learning to optimize buffer composition, boosting mRNA 20-fold, enabling cell-free transcriptomics and uncovering progressive regulatory layers in T7 phage.
|
|
Scooped by
mhryu@live.com
June 27, 4:25 PM
|
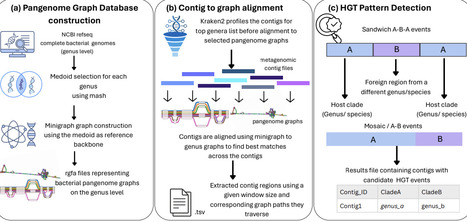
Horizontal gene transfer (HGT) shapes bacterial evolution and microbial ecosystems, yet detecting HGT within microbiomes remains a challenge due to fragmented metagenomic assemblies, reference bias, reliance on gene boundaries, and limited ability to model structural mosaicism and patterns across genomes. We present Kente, a novel pangenome graph-based framework designed for HGT detection that aligns metagenomic assembly contigs to a curated database of >600 genus-level bacterial pangenome graphs constructed using minigraph. Kente infers local taxonomic composition along contigs using alignment evidence and classifies candidate transfers using structured clade-transition topologies (e.g., A-B-A sandwich, open tips, and mosaic patterns). A complementary intra-genus module detects inter-species transfers within a single genus graph using segment-level clade annotations. Across simulated intra- and inter-genus transfer scenarios, Kente achieves higher precision and comparable recall relative to existing gene-centric microbiome HGT detection approaches while reducing false positives from fragmented assemblies. Application to real human gut metagenomes (HMP2, n = 26) demonstrates Kente's ability to detect candidate cross-lineage transfer regions in complex microbial communities. Runtime profiling shows near-linear scaling with input size, enabling efficient analysis of large metagenomic assemblies. Availability and Implementation: https://github.com/treangenlab/Kente
|
|
Scooped by
mhryu@live.com
June 27, 4:14 PM
|
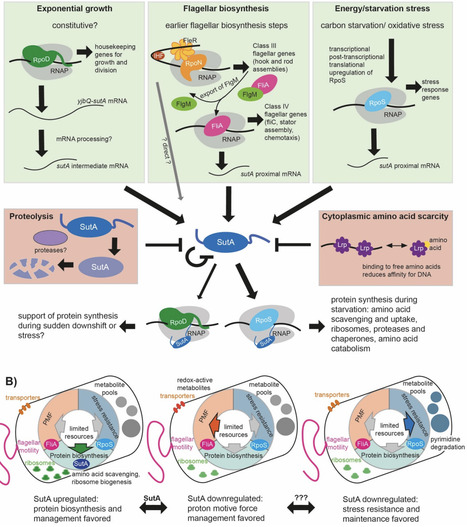
Bacteria in natural environments frequently encounter nutrient limitation leading to growth arrest and must balance the potential benefits of continuing to respond to the environment by making new proteins against the costs of depleting limited resources. We previously showed that the RNA polymerase-binding regulator SutA enhances transcription of hundreds of genes during nutrient limitation in Pseudomonas aeruginosa, suggesting that it might be part of a regulatory network facilitating limited new protein synthesis. Here, we sought to expand our understanding of this network by identifying transcriptional regulators influencing sutA expression. Using northern blotting, western blotting, and reporter assays, we found that the sigma factors FliA and RpoS, and the DNA-binding regulator Lrp, impact expression from a proximal sutA promoter during the transition to stationary phase. This constellation of regulators and the dynamics of SutA expression lead us to propose that SutA is part of a regulatory network that facilitates scavenging. Scavenging includes motility toward possible nutrient sources and uptake mechanisms for these nutrients, activities which require an investment of resources but can yield important benefits during starvation. In vitro transcription experiments, proteomic analysis and reporter assays suggest that SutA directly supports new protein synthesis driven by RpoS and indirectly supports flagellar motility, perhaps by helping maintain protein biosynthetic capacity against the metabolic costs of motility. SutA expression is controlled by multiple regulatory inputs, including negative autoregulation, and the protein appears to be short-lived. These properties are consistent with a role in supporting short, controlled bursts of gene expression during nutrient limitation.
|
|
Scooped by
mhryu@live.com
June 27, 3:28 PM
|
Bacterial systems are emerging as living technologies for sustainable manufacturing, environmental monitoring, agriculture, and health. They form biofilms, that is, living materials, which can be repurposed in biological applications due to their properties, for example, surface attachment, matrix formation, spatial organization, and stress tolerance. Advances in bioengineering have identified the benefits of biofilms for biotechnological applications. This review defines engineered bacterial biofilms (EBBs) as biofilms formed by engineered bacterial chassis and biofilm-related genes, genetically modified to perform defined biological functions. We present representative EBB engineering and application case studies and propose a life cycle framework spanning biofilm preformation, formation, and postformation. By highlighting definitions, examples, enabling tools, and translational barriers, this review provides design principles for developing reliable, safe, and application-ready biofilm technologies.
|
|
Scooped by
mhryu@live.com
June 27, 3:20 PM
|
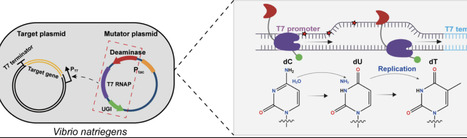
Genetic diversification serves as the foundation for in vivo molecular evolution. The fusion of a cytidine deaminase with T7 RNA polymerase (MutaT7) enables efficient diversification of DNA sequences downstream of the T7 promoter in model microbial hosts E. coli and S. cerevisiae. Vibrio natriegens, currently the fastest-growing nonpathogenic bacterium with a doubling time half that of E. coli, emerged as a promising alternative chassis for synthetic biology. Here, we report the establishment of MutaT7 in V. natriegens (dubbed VnMutaT7), achieving rapid evolution of the model protein TEM-1 to confer high-level ceftazidime resistance in just 36 h. This work establishes the potential of V. natriegens as an accelerated platform for protein engineering.
|
|
Scooped by
mhryu@live.com
June 27, 2:58 PM
|
Symbiotic nitrogen fixation reduces reliance on synthetic fertilizers and is central to agriculture and ecosystem functioning. In legumes, this process occurs in root nodules where rhizobia differentiate into bacteroids that either remain viable or undergo terminal differentiation, a strategy that can enhance nitrogen fixation but limits nodule life span. This irreversible program suits annual legumes and has evolved convergently across multiple lineages, including the inverted repeat-lacking clade (IRLC), where it is enforced by nodule-specific cysteine-rich (NCR) peptides. By contrast, perennial legumes with indeterminate nodules must sustain symbiosis over extended periods, which is incompatible with terminally differentiated bacteroids. Here, we identify a family of nodule-specific proline-glycine-rich peptides (NPGs) in Robinia that are induced upon rhizobial infection. NPGs are highly expressed in nodules, encode intrinsically disordered peptides, and accumulate in infected cells within the fixation zone. Exposure of Mesorhizobium robiniae to recombinant NPGs induces transcriptional changes associated with a fixation-related physiological state while preserving bacterial viability. These findings identify NPGs as candidate host effectors at the plant-microbe interface and point to an alternative mode of symbiont modulation in perennial legumes.
|
|
Scooped by
mhryu@live.com
June 27, 2:34 PM
|
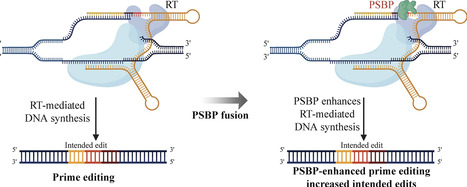
Prime editing (PE) enables precise small DNA changes yet often shows modest efficiency in human cells. Recent advances indicate that the activity of the reverse transcriptase (RT) component is a key determinant of PE performance. Here, we adapt a principle from natural polymerases by fusing a polymerase substrate binding protein (PSBP) with RT to enhance PE. In a twelve-member screen, multiple PSBPs increased editing efficiency, with a compact HMG box module, NHP6A, emerging as a representative lead fusion. NHP6A fused to RT significantly increases intended edits across substitutions, insertions, and deletions while indels remain low. The enhancement is broadly compatible with a nicking sgRNA (PE3), transient mismatch repair inhibition via MLH1dn (PE4), and structured 3′ pegRNA extensions (epegRNA). The PSBP module also synergizes with the La protein-assisted pegRNA tail stabilization (the PE7 system) for additional improvements. Using NHP6A together with La, we efficiently installed four clinically relevant alleles with high product purity. Finally, we validate that PSBP fusion can be consolidated into a single component prime editor without loss of activity. These results establish PSBP fusion as a precise route to improve prime editing outcomes and support integrating compact accessory modules into next-generation PE platforms.
|
|
Scooped by
mhryu@live.com
June 26, 3:42 PM
|
A handful of start-up firms are testing therapies that target specific epigenetic markers to treat everything from high cholesterol to a rare muscular disorder. industry
|


























omv