Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:47 AM
|
Plants have long served as natural indicators of environmental conditions, and recent advances in synthetic biology are enabling the design of engineered sentinels — living sensors that can report on abiotic and biotic stressors. This review summarizes recent advances in designing sensor plants, also called phytosensors or sentinel plants, highlighting three major strategies: (1) exploiting native promoter systems responsive to environmental cues, (2) engineering protein-based genetically encoded biosensors that detect specific molecules of interest, and (3) constructing interkingdom signaling networks between plants and microbes to extend sensing capabilities to the rhizosphere. These sense-response modules can be coupled to optical reporters (e.g., fluorescence, bioluminescence, and pigment-based) that enable remote detection via drones and satellite imaging. Continued improvements in promoter design, receptor modularity, and signal visualization technologies are driving the development of robust, field-deployable plant biosensors. Together, these innovations position engineered sensor plants as scalable, self-sustaining sentinels for real-time environmental monitoring and land management.
|
|
Scooped by
mhryu@live.com
July 2, 11:52 PM
|
Nanopore direct RNA sequencing (DRS) has transformed transcriptomics by enabling single-molecule, long-read sequencing of native RNA without the need for reverse transcription or amplification. In contrast to short-read RNA-seq and cDNA-based long-read approaches, DRS can simultaneously capture multiple RNA modifications, full-length transcript architecture, alternative splicing patterns, and poly(A) tail features within individual molecules, thereby providing an integrated view of transcriptomic and epitranscriptomic regulation. In this comprehensive review, we outline the biophysical principles underlying nanopore DRS and trace its technological evolution. We compare its performance with short-read RNA sequencing, long-read cDNA sequencing, and conventional RNA-modification mapping strategies, highlighting its advantages in isoform-resolved quantification and multilayer RNA feature integration, while also clarifying contexts in which alternative or combined approaches may be more appropriate for robust biological interpretation. We further summarize optimized experimental workflows, including library construction strategies tailored to diverse RNA biotypes (mRNA, rRNA, tRNA, circRNA, miRNA, and nonpoly(A) transcripts), as well as recommended quality-control procedures and sequencing optimization practices. Emphasizing recent computational advances and translational applications of DRS, we cover state-of-the-art algorithms for RNA modification detection, transcript reconstruction, and isoform quantification. We also propose analytical pipelines for poly(A) tail length inference and integrative frameworks that jointly analyze these regulatory layers. We distinguish direct nanopore signals from computational inferences to define confidence levels and emphasize benchmarking and orthogonal validation of readouts. Practical implementation examples are included to facilitate reproducible analysis. Finally, we highlight emerging applications of integrated DRS, including the resolution of complex transcriptomes, the characterization of coordinated epitranscriptomic regulation, and the identification of disease-associated RNA signatures. We also discuss current technical challenges and future perspectives, particularly in relation to multi-omics integration and the broader deployment of DRS in precision medicine as well as in plant and animal research.
|
|
Scooped by
mhryu@live.com
July 2, 11:34 PM
|
Artificial intelligence (AI) strategies are revolutionizing genomics by extracting complex patterns that traditional statistical pipelines are likely to miss. This mini-review aims to provide a concise overview of how AI is transforming major genomic technologies including variant calling, gene expression analysis, single-cell transcriptomics, CRISPR-Cas9 optimization, and multi-omics integration. In genome sequencing, machine learning variant callers greatly improve the accuracy and the rate at which single nucleotide and structural variants are called. In bulk RNA-Seq, AI augmented quantification, denoising, and differential expression modules complement the highly established STAR-featureCounts-DESeq2 pipeline, revealing subtle signals in big data sets. In single cell transcriptomics, deep learning approaches enhance batch correction, automate cell type annotation, and track developmental trajectories, hence clarifying cellular heterogeneity. AI-assisted guide RNA design, outcome prediction, and nuclease engineering enable more efficient CRISPR-Cas9 editing, reducing experimental cycles, and off-target effects. Finally, integrated platforms that combine genomic, transcriptomic, epigenomic, proteomic, and metabolomic layers provide an integrative view of cellular regulation and disease mechanisms. The review also covers current limitations, sparsity of data, model bias, privacy, and the need for standardized benchmarks and offers future directions in the form of interpretable models, collaborative learning, and open science practices. Together, these developments render AI an indispensable partner to unravel genomic complexity and accelerate precision medicine applications.
|
|
Scooped by
mhryu@live.com
July 2, 11:28 PM
|
CRISPR-Cas9 genome-editing efficiency is strongly influenced by the sequence composition and positional context of single-guide RNAs (sgRNAs). Although numerous deep learning–based models have been developed to predict Cas9 efficiency from sgRNA sequences, most operate as black boxes, offering limited insight into the sequence determinants underlying Cas9 activity. In addition, previous studies often overlook how the positional context of sequence motifs within sgRNAs influences their effects on Cas9 binding or cleavage. We introduce DeepCC9, an interpretable machine learning framework that combines explicit sequence feature extraction with a residual block–based deep architecture to improve interpretability and identify composition- and position-based motifs governing Cas9 genome-editing efficiency. We applied this method to multiple Cas9 variant datasets, achieving superior predictive performance compared with existing methods while enabling direct interpretation of sequence motifs and their positional effects. Our analysis uncovered 74 sequence motifs enriched or depleted at specific positions within sgRNAs and strongly associated with Cas9 efficiency, providing mechanistic insight into sequence features that influence guide performance. Together, these results establish DeepCC9 as a generalizable and interpretable framework for modeling sequence–function relationships and advancing the understanding of the sequence determinants underlying CRISPR-Cas9 genome editing.
|
|
Scooped by
mhryu@live.com
July 2, 10:49 PM
|
Lichens are symbiotic associations between a fungal mycobiont and a photosynthetic photobiont. They thrive in nutrient-poor environments; yet the mechanisms underlying their adaptation to iron limitation remained largely unknown. Here, we characterize the iron acquisition system of Xanthoria parietina, a globally distributed lichen-forming fungus associated with the microalgal photobiont Trebouxia decolorans. We demonstrate that the mycobiont produces the siderophore ferrichrome and possesses the full genetic repertoire not only for siderophore biosynthesis, but also reductive iron assimilation, iron detoxification, and regulation. The ferrichrome-synthesizing non-ribosomal peptides synthetase exhibits a lichen-specific compact architecture but retains functionality when heterologously expressed in a non-lichenized ascomycete. Transcriptomic analysis and ferrichrome quantification reveal substrate-dependent regulation of the siderophore system. Importantly, ferrichrome promotes photobiont growth independent of extracellular iron reduction, indicating direct utilization. These findings provide the functional evidence of siderophore-mediated iron acquisition in a lichen symbiosis and highlight ferrichrome as a key mediator of mutualistic nutrient exchange. The mechanisms underlying the adaptation of lichens to nutrient-poor environments are poorly understood. Here, Happacher et al. show that a globally distributed lichen fungus produces an iron-scavenging molecule that promotes growth of its algal partner.
|
|
Scooped by
mhryu@live.com
July 2, 6:04 PM
|
Anaerocellum (formerly Caldicellulosiruptor) bescii, an anaerobic, extremely thermophilic (Topt ∼78 °C) lignocellulolytic bacterium, is a promising chassis for metabolic engineering and next-generation bioprocessing. Yet, a lack of well-characterized genetic parts in A. bescii has hampered metabolic engineering efforts. Here, using a previously developed hyperthermophilic β-galactosidase reporter system, we screened a diverse panel of putative A. bescii promoter sequences, identifying promoters that drove reporter output across a broad range. For a select subset, we mapped their transcriptional start sites (TSSs) and evaluated ribosome binding site (RBS) regions using chimeric promoter constructs. By constructing truncated promoter variants, we defined functional regions within the widely used, high-expression S-layer protein promoter (Pslp) and engineered a compact 99 bp variant that retained substantial reporter activity. Finally, we demonstrated that these new promoters can be used for metabolic engineering by using two newly characterized promoters to express an established thermostable alcohol dehydrogenase from Thermoclostridium stercorarium to drive ethanol production in A. bescii. Together, this work expands and diversifies the A. bescii genetic toolkit, opening doors to future metabolic engineering efforts in this species.
|
|
Scooped by
mhryu@live.com
July 2, 5:48 PM
|
Over the past decade, protein design has evolved from a specialized discipline into a broadly accessible approach for engineering and interrogating biological systems. Despite these advances, protein design continues to be a technically challenging task, often requiring knowledge of programming to be able to use and combine the different software packages. To address this challenge, we have developed Prosculpt, an easy-to-use protein design pipeline. Prosculpt integrates RFdiffusion for backbone generation, ProteinMPNN for sequence design and multiple structure-prediction platforms (AF2, AF3, Colabfold, Boltz2). Candidate designs are evaluated using customizable Rosetta-based scoring protocols. Each project is specified through a single configuration file, enabling users with minimal computational expertise to perform sophisticated protein design tasks without writing code, while also allowing advanced users to access the full capabilities of the underlying programs. Prosculpt supports a wide range of applications, including design of symmetric homo-oligomers, design of binders, motif scaffolding, partial diffusion and fixed-backbone sequence redesign. By combining these capabilities within a single, user-friendly platform, Prosculpt provides a practical entry point to modern protein design for both novice and expert users.
|
|
Scooped by
mhryu@live.com
July 2, 5:44 PM
|
Precise spatiotemporal regulation of engineered microbes remains a critical bottleneck in synthetic biology. While ultrasound is extensively utilized for imaging and drug delivery, its translation into bacterial chassis is hindered by the lack of stringent biochemical triggers. Here, we present a rationally designed, ultrasound-responsive hybrid molecular switch based on a strict acoustic-biochemical "AND-gate". We engineered a highly sensitive split-T7 RNA polymerase system, which the dimerization and subsequent gene transcription can only be triggered in the presence of both ultrasound and a PEG-modified rapamycin. By systematically optimizing the acoustic parameters, we deployed this spatiotemporal switch to dynamically regulate a microbial consortium. With lysisE suicide protein as the output module, we achieved precise and programmable tuning of bacterial population in a co-culture system. This acoustic gating strategy may provide a robust and versatile toolkit for complex microbiome engineering and dynamic biomanufacturing.
|
|
Scooped by
mhryu@live.com
July 2, 5:13 PM
|
Current cytosine base editors (CBEs) are limited to unidirectional C to T conversions, restricting their applications. Retrons, bacterial genetic elements, encode a reverse transcriptase that generates multicopy single-stranded DNA (msDNA) by reverse transcribing specific non-coding RNA (ncRNA). This msDNA mimics Okazaki fragments during DNA replication, making retrons promising for gene editing. Here, we developed a retron-based cytosine base editor (RCBE) by fusing cytosine deaminase with reverse transcriptase (RT-CDA) within the retron system. RCBE first transcribes ncRNA, allowing RT-CDA to deaminate cytosine on the ncRNA. The modified ncRNA is then reverse transcribed into msDNA, where RT-CDA induces further cytosine deamination. This mutant msDNA introduces specific mutations into target gene sequences, enabling both C to T and G to A conversions. Using RCBE, we demonstrated accelerated molecular evolution of the rpoB gene in E. coli. High-throughput sequencing confirmed that RCBE achieves a mutation rate of up to 0.2% in regions with high GC content. Our findings establish RCBE as a versatile tool, particularly suitable for directed evolution in GC-rich regions, with broad potential applications across various bacterial and eukaryotic hosts.
|
|
Scooped by
mhryu@live.com
July 2, 4:13 PM
|
Synthetic gene circuits provide experimentally tractable systems for dissecting how genetic feedback and diffusible signals generate multicellular patterns. However, building multi-component circuits whose behavior can be quantitatively linked to module-level measurements, diffusion, and spatial boundary conditions remains challenging. Here, we designed and engineered a bacterial patterning system in which positive feedback, delayed negative feedback, and two orthogonal quorum-sensing signals are integrated in E. coli. We first implemented and characterized the feedback modules separately, measured the effective diffusion of the signals in the experimental setup, and used these data to parameterize a mathematical model. In quasi-2D bacterial lawns, the complete circuit generated self-organized spatiotemporal dynamics consisting of an sfGFP activation front followed by successive mCherry propagating pulses/traveling waves. Model-guided perturbations showed that lawn size, lawn position relative to the domain boundary, and signal degradation modulate the timing, amplitude, wavelength, and directionality of these patterns. Our work establishes a modular synthetic multicellular reaction-diffusion system in which circuit architecture, signal diffusion, and boundary-mediated signal exchange can be experimentally connected to emergent patterning dynamics.
|
|
Scooped by
mhryu@live.com
July 2, 3:51 PM
|
How microbial populations respond to repeated environmental transitions determines both their ecological fitness and their utility in biotechnological applications. Using Pseudomonas putida KT2440 equipped with fluorescent biosensors and monitored by automated flow cytometry in the Segregostat platform, we show that exposure to benzoate, a plastic-derived aromatic feedstock, progressively reduces cellular responsiveness, defined as the fraction of cells that successfully activate a gene circuit following an environmental transition. Unlike classical switching costs, which promote phenotypic diversification, benzoate suppresses responsiveness without increasing population entropy, in a concentration-dependent and circuit-independent manner tightly correlated with fitness loss. A resource allocation model incorporating the competing demands of benzoate assimilation, toxicity, and tolerance reveals that this impairment emerges from a three-way competition for limited cellular resources. Above a critical benzoate load, insufficient resources remain available to sustain the adaptive reallocation required for circuit activation. In continuous culture, a non-responsive subpopulation accumulates as a leading indicator of population collapse. Exploiting this signal, we implement a two-stage connected bioreactor system in which benzoate feeding is autonomously regulated based on real-time population structure, enabling complete substrate consumption and stable operation at otherwise destabilizing concentrations. These results establish cellular responsiveness as a quantitative population variable and demonstrate that structure-aware feedback control, acting on population composition rather than bulk physiology, provides a principled route toward autonomous bioprocesses on challenging substrates.
|
|
Scooped by
mhryu@live.com
July 2, 3:03 PM
|
Non-coding RNAs play diverse roles in a wide range of cellular processes, with their spatial structure being pivotal to their function. RNA secondary structure is a key determinant of its overall fold. Given the scarcity of experimentally determined RNA 3D structures, understanding secondary structure is vital for discerning RNA function. Currently, there is no universally effective solution for de novo RNA secondary structure prediction. Existing methods are becoming increasingly complex without marked improvements in accuracy and often overlook critical features such as pseudoknots and alternative folds. Here, we introduce SQUARNA, a new approach to de novo RNA secondary structure prediction that is suitable for both individual RNA analysis and large-scale structural searches. SQUARNA revisits the concept of base pair maximization and develops it into a stem maximization idea coupled with the widely used free energy minimization (MFE) framework. SQUARNA can predict alternative structures and handle pseudoknots of arbitrary complexity. Benchmarking shows that SQUARNA outperforms existing methods, including deep learning models, in both single-sequence and alignment-based RNA secondary structure prediction. SQUARNA seamlessly integrates sequence and alignment information with experimental data, such as residue reactivities obtained by chemical probing, as well as other structural restraints, including automated searches for Rfam database templates, G-quadruplex patterns, and protein-binding motifs. SQUARNA is available as a standalone tool at https://github.com/febos/SQUARNA and as a web server at https://larnal.imol.institute.
|
|
Scooped by
mhryu@live.com
July 2, 2:44 PM
|
Akkermansia muciniphila is a key member of the gut microbiota and plays important roles in host metabolism and health. In the colon, A. muciniphila extracts nutrients from oligosaccharide-rich mucin glycans that comprise the mucosa. However, this environment is complex and shaped by dietary inputs, microbiome metabolism, and mucin glycan composition varying across hosts, gastrointestinal regions, and physiological states. How strains of A. muciniphila integrate these nutrient signals into growth initiation and niche colonization remains unclear. Here, we compare physiological responses of a human- and mouse-derived strain of A. muciniphila, finding that dietary sugars differentially affect these isolates, suggesting host-associated tuning of metabolic capacity. In contrast, several mucin-derived sugars impose a conserved, concentration-dependent delay in growth initiation, implicating the lag phase as a critical metabolic checkpoint for growth. Genetic suppressor analysis identified sugar kinases and a component of the tricarboxylic acid cycle as genetically encoded control points linking glycan sugar exposure to the energy balance required for growth. These findings demonstrate that mucin-derived sugars function as both nutrients and metabolic stressors, regulating growth initiation. We propose that A. muciniphila employs metabolic “brakes” to coordinate growth with mucin composition, putatively linking host glycan landscapes to microbial physiology and ecological fitness within the mucus layer.
|
|
|
Scooped by
mhryu@live.com
July 2, 11:56 PM
|
Virological research has traditionally focused on individual viruses or viral families. Advances in DNA synthesis now allow large-scale construction of individual gene products, enabling systematic exploration of the virome. Here, we developed a barcoded library of ∼12,000 viral open reading frames (vORFs) from 513 viral species, which we leveraged to identify hundreds of viral regulators of cellular proliferation, MHC class I antigen presentation, and interferon signaling. Integrating results across these screens revealed unique phenotypic profiles and functional vORF modules, allowing the in-depth characterization of two previously uncharacterized viral proteins, MC162R and Yaba-like disease virus (YLDV) 151R, which impair MHC class I antigen presentation and interferon (IFN)-β signaling, respectively. Together, the viral ORFeome provides a scalable framework for dissecting viral protein function across the breadth of the virome.
|
|
Scooped by
mhryu@live.com
July 2, 11:37 PM
|
Plants operate as metaorganisms, depending on the coordinated signalling between the microbiomes of the roots (rhizosphere) and leaves (phyllosphere). This review covers recent studies that have identified rhizosphere-phyllosphere cross-talk as a crucial determinant of systemic stress resilience. Microbial metabolites, phytohormones, volatile organic compounds (VOCs), extracellular vesicles (EVs), and short RNAs (sRNAs) coordinate subterranean responses via vascular, gaseous, and molecular routes. Beneficial root-associated microbes modulate plant ethylene levels and antioxidant defense system in leaves through production of indole-3-acetic acid (IAA) and ACC deaminase activity. This causes the leaves to hold more water and chlorophyll when it is dry. In contrast, phyllosphere methylotrophs control root exudation through cytokinin-linked feedback which maintains metabolic balance. The identification of EV-encapsulated sRNAs and microbial lipopeptides as mobile nano-messengers paves way for a novel epoch in plant-microbe communication. Fungi, mycorrhizal association, and polyphagous insects are important in the regulation of nutrient fluxes and mediation of the trade-offs between nutrient acquisition and plant defense. Integrative multi-omics, isotope tracking, and synthetic community (SynCom) reconstructions now enable causal mapping of these systemic linkages. Understanding the cross-talk between different parts of the microbiome can help develop climate-resilient crops and provide a mechanistic basis for sustainable agriculture.
|
|
Scooped by
mhryu@live.com
July 2, 11:32 PM
|
DNA replication is initiated at specific chromosomal loci termed origins. In bacteria, the master replication initiation protein DnaA unwinds the origin (oriC), allowing a pair of replicative helicases to be loaded around each strand of the DNA duplex. The molecular mechanisms for managing bacterial helicase loading at oriC are unclear. Here we have investigated the role of the essential accessory helicase loader DnaB in Bacillus subtilis. By identifying and characterizing DnaB residues that are critical for its role during DNA replication initiation, we have located three necessary protein–protein interactions that DnaB makes with initiation proteins DnaA, DnaD, and DnaI. Combining single particle cryo-electron microscopy, AlphaFold3 predictions, and two-hybrid interaction analyses, the data suggests that DnaB acts as an interaction hub to orchestrate dual helicase loading at the origin. We propose a model for DNA replication initiation in B. subtilis and related Firmicutes pathogens that employ DnaB-type helicase loaders.
|
|
Scooped by
mhryu@live.com
July 2, 11:12 PM
|
Polymerase-mediated DNA synthesis is fundamental to numerous biotechnology applications, but existing programmable synthesis methods depend on exchanging DNA building blocks, thereby increasing reagent use and complicating multistep workflows. Here, we introduce the TEmperature Mediated Primer Exchange Reaction (TEMPER), a programmable platform for arbitrary DNA synthesis that operates solely through temperature control without solution exchange. TEMPER uses hairpin DNA as temperature-responsive building blocks that define specific temperature range for DNA synthesis. The temperature range is determined by the length design of the hairpin, which regulates thermodynamic interactions between DNA molecules and allows selective and sequential DNA synthesis in one-pot. We validate its versatility by developing a DNA data storage writer, a colorimetric temperature indicator, and a temperature data logger, highlighting its broad potential in nanotechnology and biotechnology applications. Polymerase-mediated DNA synthesis has numerous potential biotechnological applications. Here the authors develop TEmperature Mediated Primer Exchange Reaction (TEMPER), a programmable one-pot DNA synthesis method that stores data in DNA via temperature cycles and records thermal history.
|
|
Scooped by
mhryu@live.com
July 2, 6:07 PM
|
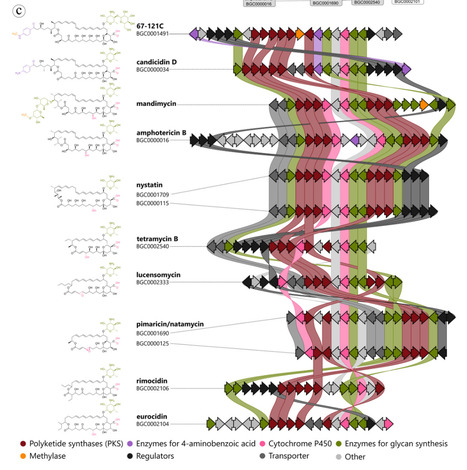
The discovery of antibiotics has historically centered on a core set of physiological targets, including cell wall synthesis, protein translation, and DNA replication. As resistance accelerates and new drug classes remain scarce, there is a growing need to expand into alternative target spaces. One such unexplored area is bacterial nutrient biosynthesis and utilization. Although their therapeutic potential is increasingly recognized, these pathways have yet to be fully integrated into antibiotic discovery pipelines, due in part to longstanding methodological biases, including the widespread use of nutrient-rich screening media that obscure nutrient-targeting activity. In this review, we highlight an overlooked subset of natural product antibiotics that inhibit nutrient metabolism. We consolidate 73 compound classes primarily retrieved from the Dictionary of Natural Products and categorize them into four mechanistic classes: biosynthesis inhibitors, antimetabolites, pro-antimetabolites, and riboswitch inhibitors. Many display whole-cell activity, including against Gram-negative pathogens, and reveal underappreciated structural and functional diversity. Recent advances in defined media design, genome mining, and synthetic biology make these compounds more readily accessible for systematic re-evaluation and optimization. Nutrient pathway inhibitors offer a source of novel antibiotic scaffolds and a foundation for therapeutic strategies such as drug potentiation and resistance reversal. Reintegrating these compounds into discovery pipelines can help diversify antibacterial options and address pressing resistance challenges.
|
|
Scooped by
mhryu@live.com
July 2, 5:58 PM
|
Biomolecular condensates formed through liquid–liquid phase separation (LLPS) compartmentalize biochemical reactions without enclosing membranes, enabling spatiotemporal control over diverse cellular processes. Engineering genetically encoded proteins that phase separate in response to defined chemical inputs remains a central challenge for synthetic biology. Here, we report a coiled-coil peptide polymer, M1, that undergoes cofactor-dependent condensation both in vitro and in E. coli. M1 is an ABA triblock construct comprising two terminal helical domains connected by a flexible, intrinsically disordered linker. The terminal domains are derived from a heme-responsive coiled-coil motif that is destabilized in the apo state but assembles into a four-helix bundle upon metalloporphyrin coordination. We demonstrate that M1 forms condensates exclusively in its cofactor-bound state, both in vitro and in cells. In E. coli, these intracellular condensates accumulate at the cell poles in a concentration-dependent manner. Depletion of cellular heme biosynthetic capacity suppressed condensate formation, which was rescued by supplementation with the heme precursor δ-aminolevulinic acid (δ-ALA) and iron, consistent with metalloporphyrin coordination triggering assembly. The condensates retain peroxidase activity characteristic of heme-containing proteins and catalyze the oxidation of Amplex Red to resorufin both in vitro and in living cells. These results establish metalloporphyrin binding as a molecular switch for condensate biogenesis in a structured peptide polymer, directly coupling cofactor coordination, mesoscale assembly, and catalytic function within a single designed system.
|
|
Scooped by
mhryu@live.com
July 2, 5:46 PM
|
Extracellular protease activity plays key roles in cell and tissue behavior, processing cell surface proteins, ligands and the extracellular matrix. Extracellular proteases can be subject to complex post-translational regulation, yet it remains challenging to quantify their activity in single cells over time. We present eNRGies (engineered neuregulin reporters as generalized indicators of extracellular shedding): modular, genetically encoded protease biosensors that translate extracellular cleavage into nuclear translocation of an intracellular (fluorescent) protein domain. We optimize the platform to report on protease activity in single cells on a timescale of minutes, and show it can be applied to soluble and cell-surface proteases including TEV protease, enterokinase, Factor Xa, MMP-9, and the sheddase ADAM17. We find ADAM17 activity can be transiently activated during mitosis and exhibit complex dynamics following EGF receptor stimulation. eNRGies biosensors enable observation of extracellular protease activity with high spatiotemporal resolution, and could be applied as synthetic biology scaffold to translate protease activity into customized cellular responses.
|
|
Scooped by
mhryu@live.com
July 2, 5:19 PM
|
Optogenetic methods are powerful tools for synthetic biology, allowing light to control cellular processes. While most bacterial optogenetic systems regulate gene expression at the transcriptional level, relatively few enable post-translational control, which can provide faster and growth-independent regulation of protein activity. Here, we describe the development of a post-translational optogenetic tool in E. coli using the Mesoplasma florum Lon (mfLon) protease, an AAA+ protease that is orthogonal to native E. coli degradation machinery. To engineer a light-responsive mfLon, we constructed a large library in which a blue-light responsive domain, Avena sativa LOV2, was introduced into nearly every codon position in the protease using an unbiased molecular approach. We screened 726 mfLon-LOV variants using fluorescence-activated cell sorting and multi-round enrichment campaigns. We identified a novel dark-active variant (mfLon-LOV-534) that degrades target proteins in the dark and is inactivated upon blue-light exposure. Characterization of this variant demonstrates that its proteolytic response can be tuned by varying blue-light intensity and transcriptional expression levels. Furthermore, we show that mfLon-LOV-534 can degrade a target protein in both exponential and stationary growth phases, which addresses the limitations of division-based protein dilution. This work establishes a scalable approach to engineering allosteric control in complex multimeric enzymes and provides a foundation for orthogonal, growth-independent control of protein stability in synthetic circuits.
|
|
Scooped by
mhryu@live.com
July 2, 4:23 PM
|
Bacterial colonies grow within microenvironments that they continuously reshape through nutrient uptake, metabolism and mechanical interaction. However, in colonies carrying engineered gene circuits, how these self-generated environmental changes feed back on gene expression to produce spatiotemporal organization remains poorly understood. Here we show that growth and gene expression are dynamically coupled during the maturation of founding colonies, with growth-driven environmental changes organizing gene expression into traveling waves. By combining quantitative time-lapse microscopy, mathematical modeling and image-based parameter inference, we demonstrate that edge-dominated colony expansion consistent with mechanical constraints on growth is followed by density-dependent growth arrest in which the total area occupied by colonies does not converge to a fixed carrying capacity of the shared growth environment. We then find that colonies form two distinct traveling waves of gene expression. An intra-colony wave emerges only after growth arrest and is observed in both constitutive genes and distinct regulated circuit architectures, indicating that it is not a result of circuit topology. Our observations are consistent with nutrient depletion during growth followed by subsequent recovery after growth arrest. At later times, an inter-colony wave emerges that is consistent with a colony-produced diffusible factor spreading through the shared medium. Together, these findings reveal that the colony environment is not a passive background but an active and intrinsic component of spatiotemporal gene regulatory dynamics, in which self-generated environmental feedback couples mechanical constraints, nutrient dynamics and gene expression across spatial scales.
|
|
Scooped by
mhryu@live.com
July 2, 4:02 PM
|
Precise regulation of protein synthesis is fundamental to cellular homeostasis and remains a primary target for synthetic biology applications. However, the non-linear relationship between mRNA abundance and protein levels presents complexities that poses challenges for predictive engineering. Here, we present TRIM, a Transformer-based RNA Inference Model that leverages full-length mRNA sequences and multi-omics data to predict translation efficiency. By employing a Parallel Expert Mixer, TRIM achieves robust prediction accuracy (R2 ≥ 0.8,Pearson r ≥ 0.9). Trained on multimodal data from massive Saccharomyces cerevisiae isolates, TRIM demonstrates outstanding biological interpretability, helping to decipher complex translational patterns such as synergistic effects between bases, sequence-dependent codon preference in different stages, and distinct attention on key secondary structures. These results indicate that the integration of multi-omics data with holistic sequence modeling can effectively decode the cis-regulatory grammar of translation as well as providing a scalable and interpretable generative framework for future synthetic biology engineering.
|
|
Scooped by
mhryu@live.com
July 2, 3:42 PM
|
The mutational spectrum is an increasingly important molecular phenotype that quantitatively describes mutagenesis in a given gene and species, enabling future comparative analyses to reveal differences in underlying mutagenic processes, whether internal, such as DNA repair processes, or external, such as ecological niches and conditions. Mutation accumulation experiments, although time-consuming and costly, remain the standard approach for reconstructing bacterial neutral mutation spectra. Here, we present BacNeMu, a phylogenetically informed pipeline that reconstructs neutral mutational spectra of bacterial genomes using open databases GTDB, AnnoTree and KEGG Orthology, building on previously developed NeMu pipeline. BacNeMu reconstructs mutation spectra that closely match mutation accumulation experiments results while requiring substantially less time, enabling comparative analyses across diverse bacterial taxa. Applied to obligate aerobes and anaerobes, BacNeMu recovered the expected excess of T:A>C:G transitions, consistent with oxidative-damage-associated mutational patterns previously described in mitochondrial genomes and yeast single-strand. We further asked if any other ecologic factors influence a mutational spectrum. As a pilot we compared three species living under different temperatures: one strong thermophile - Thermotoga maritima, one psychrophile - Clostridium algidicarnis, and one with intermediate temperature tolerance - Psychrobacter sanguinis. In the thermophile, the relative frequency of T:A>C:G substitutions was higher than in the psychrophile, consistent with the hypothesis that GC-biased mutagenesis contributes to thermal adaptation, although C:G>T:A transitions predominate across all three species. BacNeMu provides a rapid, phylogenetically informed framework for generating biologically meaningful mutation spectra from open databases.
|
|
Scooped by
mhryu@live.com
July 2, 2:47 PM
|
Neisseria gonorrhoeae is a common Gram-negative pathogen with increasing resistance to all recommended antibiotics. There is a critical need to improve the efficiency of the antibiotic hit discovery process to replenish the drug development pipeline. Here, we show that deep learning models can augment high-throughput screens to identify readily available molecules with narrow-spectrum activity against difficult-to-treat strains of N. gonorrhoeae. We phenotypically tested 38,650 small molecules for N. gonorrhoeae growth inhibition to train a predictive graph neural network (GNN) model. We benchmarked the model’s performance against other architectures, including a large language model, and found that GNNs more accurately identify active, drug-like molecules that are structurally distinct from the training set and known antibiotics. Using the model to virtually screen ~6 million compounds, we identified 213 compounds for experimental validation and found that 83 (39%) inhibited N. gonorrhoeae growth. Two of these compounds were structurally dissimilar to existing antibiotics, maintained potency against multidrug-resistant N. gonorrhoeae strains in vitro, exhibited promising selectivity indices, and were rapidly bactericidal with low frequencies of resistance. Proteomic studies revealed their distinct mechanisms of action, with one compound targeting alanine racemase, an enzyme involved in the essential process of peptidoglycan synthesis. Furthermore, the compounds showed early promise in reducing N. gonorrhoeae titers in a human vagina-on-a-chip infection model and a mouse vaginal infection model. Our work establishes the deep learning–enabled discovery of selective antibacterial compounds against N. gonorrhoeae as a much-needed hit discovery tool to address the growing crisis of antimicrobial resistance for this pathogen.
|