 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:23 AM
|
Advances in structural biology have improved our understanding of the relationship between protein structure and function, while also confirming a widely applicable principle: protein domains with highly conserved three-dimensional folds can perform radically disparate biochemical functions. To gain insight into this structural enigma, we mapped the energetic landscapes of a family of bacterial transcription factors and their anciently diverged structural homologues, the periplasmic binding proteins. Using hydrogen exchange–mass spectrometry, bioinformatics, X-ray crystallography and molecular dynamics, we uncovered an unexpected contrast: despite binding the same sugars, the two families have evolved unique ‘energetic blueprints’ to support their distinct functional requirements. To test if differences in ensemble energies have functional consequences, we rationally redesigned the protein fold for tunable ligand-driven transcriptional responses. Strikingly, energy-driven protein engineering produced synthetic transcription factors with the theoretically anticipated ligand-induced transcriptional outputs. Thus, decoding energetic blueprints among conserved protein folds provides diverse functional adaptations, paves an alternative roadmap for protein design, and offers a distinct approach for engineering challenging drug targets. Proteins sharing a common fold can evolve strikingly different functions, but the underlying energetic logic is often hidden. Now, hydrogen exchange–mass spectrometry reveals conserved energetic ‘blueprints’ that distinguish Venus flytrap transcription factors from transport proteins, uncovering molecular switches for allostery and enabling rational tuning of ligand sensitivity.
|
|
Scooped by
mhryu@live.com
June 6, 11:24 PM
|
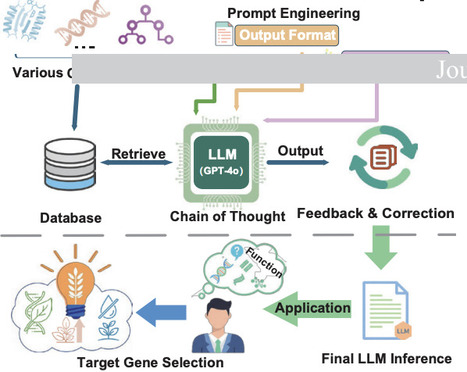
GENIUS-LLM takes a plant gene ID as input, retrieves associated multi-omics data including expression profiles, GO/KEGG annotations, TWAS results, co-expression networks, and sequence homology, and synthesizes that evidence into a written report stating what the gene likely does and why, with each functional claim linked to the specific evidence supporting it. Direct functional evidence is weighted more heavily than indirect signals, so the reasoning reflects evidence quality. annotation
|
|
Scooped by
mhryu@live.com
June 6, 11:13 PM
|
Microbial cultivation optimization remains labor-intensive and inefficient, requiring extensive experimental screening to identify suitable growth conditions. Traditional one-factor-at-a-time approaches are particularly ineffective for exploring complex, multidimensional nutrient parameter spaces. We present MicroGrowAgents, an AI-driven, agent-based system that automates the design of optimized growth media through integration of knowledge graphs, metabolic modeling, and optimal experimental design. The system employs 28 specialized agents and 50 skills that query structured biological knowledge (KG-Microbe: 864,363 validated species), mine literature evidence (245+ papers), perform genome-guided design (57 genomes, 667,000+ annotated features), and generate statistically optimal experimental designs using the MaxPro algorithm. We applied the approach to Methylorubrum extorquens AM1 by cultivating 70 designed conditions in quadruplicate and assessing three concurrent objectives: biomass (OD600 at 740 nm), redox activity (Abs590 Biolog proxy), and lanthanide uptake (residual Nd measured by arsenazo III). Monte-Carlo resampling of the replicate-level uncertainty (1000 iterations) identified a single stable Pareto-optimal medium, MPOB_058 (membership frequency 0.99), together with two borderline candidates and six rare appearers, providing a robust anchor set for subsequent rounds of design-build-test-learn. The integration of chemical similarity search (208,000+ embeddings), metabolic gap analysis, and multi-modal reasoning enables evidence-based hypothesis generation that reduces experimental burden while accelerating discovery of growth-promoting conditions. MicroGrowAgents provides complete provenance tracking with cryptographic checksums and 90.5% literature citation coverage, advancing reproducible, data-driven approaches to microbial cultivation.
|
|
Scooped by
mhryu@live.com
June 6, 11:01 PM
|
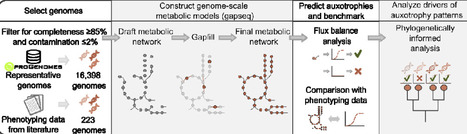
Amino acid auxotrophy, the loss of biosynthetic potential for an amino acid, is a highly prevalent feature of microbial life, yet the evolutionary processes driving its distribution in nature remain unclear. Here, we sought to determine whether auxotrophy emerges as an independent response to nutrient availability or as part of a broader transition in genomic lifestyle. By first predicting auxotrophies through genome-scale metabolic models and flux-balance analysis, and subsequently applying phylogenetically informed statistical frameworks, we tested the predictions of the Black Queen Hypothesis, which posits that auxotrophy evolves as a dynamic response to metabolite availability, and disentangled the signal of convergent environmental selection from the background of shared evolutionary history. This approach reveals that phylogeny is the dominant predictor of auxotrophy, vastly outweighing environmental context. Beyond correcting for ancestry, our analysis uncovered a previously obscured signal of consistent, co-occurring genomic changes: auxotrophies do not occur as independent losses, but rather as part of a broader reorganization of the genome. We show that auxotrophies co-occur significantly more often than predicted by chance and that the probability of loss scales with the number of other auxotrophies. These findings reveal auxotrophy as a feature of a conserved, phylogenetically entrenched lifestyle rather than a transient response to nutrient availability - inconsistent with the Black Queen Hypothesis and instead aligning with genome streamlining as the overarching driver of metabolic dependency in natural microbial systems.
|
|
Scooped by
mhryu@live.com
June 6, 5:08 PM
|
Temperature is a fundamental constraint on life. Most organisms adapt to temperature change through complex regulatory and stress-response systems. Whether such adaptation is possible in a genomically minimal cell remains unclear. Here, adaptive laboratory evolution was performed on the minimal genome bacterium JCVI-syn3B, shifting growth from 37℃ to 25℃, a temperature at which the original strain cannot grow. After 40 serial passages, the evolved strains exhibited robust growth at 25℃. Multi-omics analyses revealed the adaptation mechanisms including increased mRNA turnover, enhanced DNA unwinding and stabilization of replication intermediates, elevated glycerolipid synthesis, and upregulation of division proteins associated with Z-ring assembly. Whole-genome transplantation was performed to distinguish genetic or non-genetic contributions, demonstrating that the cold-adapted phenotype was largely genetically encoded. These results indicated that even a minimal cell retains substantial evolvability and highlight its potential as a tractable research platform for cellular and evolutionary biology.
|
|
Scooped by
mhryu@live.com
June 6, 4:13 PM
|
Far-red light photoacclimation enables some cyanobacteria to survive in white-light-depleted environments by extending the red limit of photosynthesis. In far-red Photosystem II, paralogous subunits replace their canonical counterparts, allowing the incorporation of some chlorophyll f molecules and one chlorophyll d that are red-shifted and spectrally distinct from the chlorophyll a manifold, and from each other. Here, we present a comparative study of far-red Photosystem II from Chroococcidiopsis thermalis PCC 7203 and Calothrix sp. NIES-3974. In C. thermalis, the cryo-electron microscopy structure reveals the far-red-exclusive subunit, PsbH2’, which forms part of a chlorophyll f binding site. We also assign four chlorophyll f sites using sequence comparisons and electrostatic potential analyses. In Calothrix, psbH2’ is absent, and the same analyses show that only two of these chlorophyll f sites are present. Comparative phylogenetic, structural, and spectroscopic analyses allow the assignment of specific wavelengths to all the red-shifted chlorophylls. This provides the framework needed to model excitation energy transfer in far-red Photosystem II, and to understand the conserved features that allow survival under far-red light. Some cyanobacteria do oxygenic photosynthesis with near-infrared photons using specialized pigments. Here, the authors identify their positions and colors by combining phylogenetic, spectroscopic, and structural methods.
|
|
Scooped by
mhryu@live.com
June 6, 3:11 PM
|
Synthetic biology is an interdisciplinary field that integrates knowledge and techniques from modern biology and many other disciplines to design and construct novel biological systems or to modify existing life forms. Its core technologies include gene editing (e.g., CRISPR/Cas9), DNA assembly, in vivo directed evolution, and integration with artificial intelligence. The development of these technologies has greatly advanced the application of synthetic biology in medicine. In disease diagnosis, engineered bacteria have shown considerable promise. They can be designed to sense disease-specific signals and produce detectable reporter outputs, thereby establishing new paradigms for early diagnosis and real-time disease monitoring. For example, bacteria engineered via synthetic biology have been developed as "living sensors" to detect disease biomarkers. In therapeutic applications, synthetic biology offers a fresh perspective on using microorganisms to treat diseases. Researchers can design and construct microorganisms with tailored functions for targeted drug delivery, immunotherapy, and microbiome modulation. These applications not only improve the precision and efficacy of treatments but also offer innovative solutions to overcome the limitations of conventional therapeutic approaches. However, despite their considerable potential, the clinical translation of engineered bacteria still faces numerous challenges, such as ensuring stable in vivo colonization, controlling immunogenicity, standardizing large-scale production, and establishing robust regulatory and ethical frameworks. This review summarizes engineering strategies aimed at enhancing the safety and efficacy of bacterial therapies, with the goal of optimizing bacterial functions and expanding their potential in diagnostics and precision medicine.
|
|
Scooped by
mhryu@live.com
June 5, 5:24 PM
|
Three different cultures of cyanobacteria: Synechocystis sp., Leptolyngbya sp., and a co-culture of both Synechocystis sp. and Synechococcus sp. were tested for their polyhydroxybutyrate (PHB) production potential under diverse conditions. High-throughput screening in a multi-well plate, using Nile blue fluorescence to estimate PHB accumulation, enabled simultaneous testing of multiple parameters without complex setups and faster PHB estimation compared to conventional gas chromatography. Fluorescence results indicated that the best conditions for PHB accumulation were nitrogen depletion under darkness at 30°C with acetate supplementation after 4 days of glycogen accumulation. Finally, these optimal conditions were validated in single-batch proof-of-concept photobioreactors (1 L working volume). The scale-up proved successful, yielding 7% PHB dry cell weight (dcw) for Synechocystis sp. and a promising 13% PHB dcw for Leptolyngbya sp. In addition, scale-up experiments in 1-L photobioreactors demonstrated that a dedicated glycogen pre-accumulation step is unnecessary, as PHB can be efficiently synthesized directly from acetate.
|
|
Scooped by
mhryu@live.com
June 5, 5:10 PM
|
Indigoidine is a blue pigment biosynthesized by a single-module Non-Ribosomal Peptide Synthetase (NRPS) using L-glutamine as substrate. Despite its potential as a colorimetric reporter, no such system has been established from it to date. We used a recently characterized interdomain fusion site located between its adenylation (A) and thiolation (T) domains to develop the Indi2GO system, which provides a naked-eye detectable and quantitative optical readout of transient and covalent protein-protein-interaction (PPI) in living cells. Indi2GO enables high-throughput benchmarking and optimization of PPI tools in a standard 96-well plate reader format, without requiring exogenous substrates, specialized equipment or complex analytical workflows. We demonstrate its broad applicability with three widely used protein-protein interaction tools: SYNZIPS, inteins, and the SpyTag:SpyCatcher system. We used Indi2GO to validate novel SYNZIP pairs, which we used in NRPS engineering, highlighting its applicability for the development of novel PPI-mediating tools in the context of NRPS engineering and synthetic biology.
|
|
Scooped by
mhryu@live.com
June 5, 3:19 PM
|
Nanoparticles are now central to many drug and vaccine delivery strategies, but most require reformulation for each application. Bacteriophage‑derived nanoparticles offer a genetically encoded, structurally defined, and modular alternative. This review organizes recent advances along three tunable axes — scaffold, surface, and cargo — and highlights hybrid phage–polymer/lipid/inorganic constructs that expand stability, targeting, and loading. We survey applications from multivalent vaccines and oversized gene transfer to precision microbiome editing, and outline translational hurdles. Phage-derived products are approaching translation, with virus-like particle-based vaccines and CRISPR-enhanced antimicrobial phages in clinical trials. Finally, we preview emerging opportunities, including AI‑guided capsid and receptor‑binding protein design, cell‑free phage synthesis, and standardized ‘reference’ phage chassis that can be combined with traditional nanoparticles, positioning phage nanoparticles as reusable, plug-and-play nanomedicines.
|
|
Scooped by
mhryu@live.com
June 5, 2:38 PM
|
In contrast to the rod shape at 37°C, the morphology of E. coli cells at temperatures just above the minimum temperature of growth is small rods. A study was initiated to determine the requirement of nucleoid-associated protein FIS for growth and genome compaction in the small rods at low temperature. Growth and nucleoid staining analyses revealed that the fis null mutant displayed decreased growth and initially formed filaments containing decondensed nucleoids at 12°C, indicating that FIS facilitates production of small rods with condensed nucleoids at low temperature. However, characterized by biphasic growth at low temperature, the fis null mutant exhibited increased growth, cell division, and nucleoid condensation following an acclimation phase. Therefore, the absence of FIS with nucleoid decondensation leads to an adaptation mechanism, termed FIS Null Adaptation Response, that causes a shift towards nucleoid condensation resulting in genome compaction in small rods. Furthermore, overproduction of the HsIVU protease suppressed the cold-sensitive phenotypes of the fis null mutant indicating that degradation of a natural substrate of the protease alleviates the requirement of FIS at low temperature. In addition, null mutations of genes encoding natural substrates of HsIVU (exoribonuclease RNAse R, and cell division inhibitor SulA) were identified as extragenic suppressors of the fis null mutation.
|
|
Scooped by
mhryu@live.com
June 5, 2:26 PM
|
Simultaneously introducing diverse genomic edits remains a challenge in crop genome engineering. Here we describe a twin prime editing-based knockout (TKO) system that installs stop codon clusters (SCCs) for precise translational termination with minimal in-frame mutations. TKO achieves knockout efficiencies of up to 70.5%, 58.6% and 75.1% in rice, maize and wheat protoplasts, respectively, and produces heritable knockout alleles in 96.8% of regenerated rice plants. In hexaploid wheat, TKO outperforms Cas9 4.2-fold in generating triple-homolog knockouts, largely by reducing in-frame mutations. Orthogonal TKO editors with sequence-divergent SCCs enable simultaneous knockout of up to ten genes without cross-interference. Integration of TKO with conventional prime editing establishes TRIM1 (TKO editor-enabled gene rupture and development of integrated multitype genome modification system) for simultaneous knockout and precise editing, achieving a 22.8% coediting of four genes in rice. TRIM2 extends this capacity to kilobase-scale modifications through a prime editor–recombinase system, enabling a 4.9-kb insertion (1.2% efficiency) and gene knockout (up to 79.8%) in protoplasts. Plant genome editing is multiplexed with twin prime editing.
|
|
Scooped by
mhryu@live.com
June 5, 12:17 AM
|
Understanding protein functions is crucial for interpreting microbial life; however, reliable function annotation remains a major challenge in computational biology. Despite significant advances in bioinformatics methods, ~30% of all bacterial and ~65% of all bacteriophage (phage) protein sequences cannot be confidently annotated. In this review, we examine state-of-the-art bioinformatics tools and methodologies for annotating bacterial and phage proteins, particularly those of unknown or poorly characterized function. We describe the process of identifying protein-coding regions and the systems to classify protein functionalities. Additionally, we explore a range of protein annotation methods, from traditional homology-based methods to cutting-edge machine learning models. In doing so, we provide a toolbox for confidently annotating previously unknown bacterial and phage proteins, advancing the discovery of novel functions and our understanding of microbial systems.
|
|
|
Scooped by
mhryu@live.com
June 6, 11:28 PM
|
Sequence-unrelated but structurally similar (SUSS) effector families represent a distinctive evolutionary strategy among plant pathogen virulence proteins. Within families such as MAX, LARS and RALPH effectors, individual proteins maintain nearly identical three-dimensional folds despite minimal sequence identities, whilst targeting functionally diverse host cellular processes. This decoupling of structural conservation from functional specificity challenges traditional precepts of the classic structure–function paradigm and reveals how pathogen effectors exploit stable protein scaffolds as platforms for rapid functional diversification through extreme sequence variation. Comparative structural analyses suggest that surface frustration, regions of local energetic instability essential for fold flexibility, may be conserved across SUSS family members despite sequence divergence. This conservation creates potential vulnerabilities that could be exploited for resistance engineering. Rather than targeting individual effector-host interactions, frustration-guided design of molecular sponges, synthetic integrated domains or proteome degradation warheads could potentially neutralise entire SUSS effector families. This review explores the mechanisms of functionalisation by SUSS effectors and suggests strategies combining structural genomics, surface frustration analysis and AI-driven protein design for developing broad-spectrum resistance against major classes of plant pathogen effectors.
|
|
Scooped by
mhryu@live.com
June 6, 11:15 PM
|
Modification of the gut microbiota by beneficial microbes can enhance an organism’s lifespan, giving rise to the concept of probiotics. Probiotics are live microorganisms that provide health benefits when taken in sufficient amounts. Owing to their outstanding health benefits, probiotics have experienced rapid expansion and gained interest for the development of new applications. The exploration of microbial applications via genetic modification is currently of great interest to researchers. Genetic engineering using CRISPR-Cas system has received considerable attention and has established applications. Owing to these enhanced properties, the CRISPR-Cas system is currently used in medicine, agriculture, food, and biotechnology. Considering the adaptive immune system in bacteria, this genetic tool is used to alter the microbial genome. Lactic acid bacteria (LAB) are widely recognized for their probiotic potential, and over 40% of LAB species contain the CRISPR-Cas system. The rising demand for probiotics and their expanding applications necessitate the enhancement of their existing characteristics. The CRISPR-Cas system, recognized for its precision, accuracy, and speed, has enabled researchers to modify the genomes of probiotics, thereby enhancing their beneficial attributes. This system can enhance probiotic properties through additive, subtractive, or modulatory mechanisms. Various approaches have been developed to improve probiotic functionalities using the CRISPR-Cas system, such as substituting slow promoters with efficient alternatives, eliminating undesirable components, boosting metabolism, and increasing tolerance levels. Furthermore, CRISPR-engineered probiotics have emerged as next-generation probiotics with enhanced properties and advanced applications across diverse fields, including the food, medicine, agriculture, and pharmaceutical sectors.
|
|
Scooped by
mhryu@live.com
June 6, 11:11 PM
|
Regulatory evolution can alter phenotypes, but cis- and trans-regulatory mechanisms may also diverge extensively while total transcript abundance remains stable. Comparisons of parental expression with allele-specific expression in F1 hybrids provide a framework for separating cis- and trans-regulatory effects because both parental alleles are measured in a shared trans-regulatory environment. Here, we analyzed RNA sequencing data from Saccharomyces cerevisiae, Saccharomyces paradoxus, and their F1 hybrid. Regulatory divergence was widespread, with 61.3% of tested orthologs showing significant divergence in at least one cis or trans component. However, hybrid expression remained largely conserved, with 81.6% of genes not significantly different from either parent. Compensatory cis-trans divergence predominated over reinforcing divergence, consistent with widespread buffering of transcript abundance. To connect genome-wide patterns to mechanism, we analyzed the strongly cis-diverged locus LYS2 and found species differences in promoter architecture, including an S. cerevisiae-specific AT-rich insertion, altered spacing among candidate regulatory features, and a promoter-proximal TATA-like element unique to S. cerevisiae. Sequence-based nucleosome prediction suggests that these differences create a broader promoter-proximal nucleosome-depleted region in S. cerevisiae than in S. paradoxus. We also quantified allele-resolved intron retention and found that splicing was broadly conserved, with only rare locus-specific hybrid-associated shifts. Together, these results show that regulatory divergence is widespread but often buffered in the hybrid, whereas post-transcriptional divergence is comparatively limited.
|
|
Scooped by
mhryu@live.com
June 6, 5:20 PM
|
Nitrogenase is the only enzyme capable of biological nitrogen fixation and a major target for sustainable agricultural engineering, yet its functional and structural complexity has made it difficult to identify regions that can be modified without compromising activity or assembly. Here, we use structural evolution to identify a lineage-specific N-terminal extension in NifK as a candidate engineering target within the MoFe protein interface. By generating variant libraries exceeding 9,000 members and evaluating them through deep mutational scanning, diazotrophic growth assays and in vitro characterization to map the sequence-function landscape of this interface across variable conditions. We show that NifK extension is required for nitrogenase activity while remaining broadly tolerant to mutation, revealing a sequence-function landscape that is both flexible and constrained. A subset of residues at the NifD-NifK interface are critical for maintaining complex stability. Structural analyses indicate that the extension stabilizes the MoFe complex via co-evolved electrostatic interactions, and targeted mutations can improve both enzymatic activity and thermostability. These findings identify the NifK extension as a tunable interface, providing a strategy for engineering more robust nitrogenases.
|
|
Scooped by
mhryu@live.com
June 6, 4:26 PM
|
Recently, high-throughput experimental techniques have propelled improvements in deep learning-based prediction of mutation effects on protein stability. However, leading stability predictors still struggle to predict the combined effect of multiple mutations and prefer mutations that negatively impact other properties, including expressibility. To mitigate these limitations, we apply Low-Rank Adaptation (LoRA) to specialize ESM3 for stability prediction by fine-tuning on the Megascale protease susceptibility dataset, developing a novel dual-perspective inference mechanism to provide explicit mutant context information. ESM-Mutant Stability Ranker (ESM-MSR) significantly exceeds all contemporary methods tested on the prioritization of stabilizing mutations (ΔNDCG@96 >= +0.12), double mutant ranking (Δρavg >= +0.068) and direct epistasis ranking (Δρavg >= +0.164) within the Megascale test set. Further, it generalizes effectively to heterogeneous thermostability benchmarks, consistently matching or exceeding current approaches across our comprehensive suite. Finally, a single parameter σ enables tunable control of the model′s compromise between stability and more general sequence fitness, leading to state-of-the-art performance in the Human Domainome 1 benchmark (Δρavg = 0.573) at σ = 0.5, demonstrating the broad applicability of ESM-MSR as a protein engineering tool.
|
|
Scooped by
mhryu@live.com
June 6, 3:48 PM
|
Developing artificial synthetic pathways for converting one-carbon compounds into value-added chemicals represents a promising strategy for carbon-neutral manufacturing. Enzymatic C–C bond formation plays a central role in carbon-chain extension and structural diversification. Fructose-6-phosphate aldolase (FSA) has been employed in in vitro multienzyme systems for producing starch and sugars from methanol. However, its atomistic catalytic mechanism has remained unclear, limiting rational enzyme engineering. Here, we elucidate the aldol reaction mechanism between dihydroxyacetone (DHA) and glyceraldehyde-3-phosphate (GALP) catalyzed by FSA using QM/MM calculations, identifying the Schiff-base/iminium formation step as the rate-limiting step in the aldol condensation. Guided by this mechanism, we engineered FSA variants with a 34-fold increase in catalytic efficiency compared with the wildtype. We further integrated the engineered FSA into a designed in vitro cascade converting methanol to mannitol, achieving a high yield of 88%. Collectively, these mechanistic insights and improved biocatalysts expand the toolkit for green enzymatic C–C bond formation and one-carbon utilization.
|
|
Scooped by
mhryu@live.com
June 6, 3:09 PM
|
Signaling-based anti-bacteriophage systems such as CBASS and Thoeris synthesize infection-triggered nucleotide signals that activate antiphage effectors. However, the phage features sensed by these systems and the mechanisms phages use to evade signaling immunity remain poorly understood. Here, studying clinically relevant Pseudomonas aeruginosa phages from the Migulavirinae family, we show that closely related phages encode subtle allelic variation in side tail fiber proteins that determine sensitivity to type II Thoeris. In parallel, these same phages encode an anti-defense hotspot that contains three adjacent genes that are each sufficient to facilitate phage evasion of both CBASS and Thoeris defenses, counter-balancing the activating proteins. Comparative analysis of this anti-signaling hotspot across the broader family of related N4-like phages uncovered a new Thoeris anti-defense (Tad) protein that sponges NAD-derived molecules (e.g. gcADPR) and exhibits sequence and structural similarity to a poorly characterized nucleotide-binding region of the human ryanodine receptor. Together, these findings reveal how the balance between immune activation and antagonism shifts phage outcomes and reveals a surprising similarity between a phage molecular sponge and an important human protein.
|
|
Scooped by
mhryu@live.com
June 5, 5:16 PM
|
Predicting protein-ligand complex structures is a central challenge in drug discovery. While recent co-folding models such as AlphaFold-3 achieve accurate structure prediction, they fail to generalize to underexplored binding interfaces - systematically misplacing ligands, particularly for allosteric or structurally novel targets. To address this gap, we present ACER (A daptive Co-folding via pocket E xploration and pose R anking), a training-free framework that (a) enables co-folding models to systematically explore alternative binding pockets, and (b) leverages the discovered pockets to increase pose accuracy. Our method enables the efficient discovery of non-prevalent pockets without prior expert knowledge. ACER improves pocket discovery and pose accuracy on allosteric targets and structurally novel complexes, successfully modeling binding interfaces that are under-represented or absent from the training set. Our results demonstrate how improved sampling dynamics enhance the generalizability of co-folding models without retraining.
|
|
Scooped by
mhryu@live.com
June 5, 4:56 PM
|
The widespread emergence of antibiotic-resistant pathogens poses a significant global health challenge and underscores the need for novel approaches to accelerate antimicrobial discovery. Antimicrobial peptides (AMPs) have gained attention as promising candidates due to their broad-spectrum activity, including efficacy against multidrug-resistant bacterial strains. SAJO-2, an antimicrobial peptide developed by Sarojini and colleagues, features a tryptophan zipper-like motif incorporating a central d-Phe-2-Abz unit, where 2-Abz functions as a conformationally constrained β-turn-inducing peptidomimetic scaffold. Modification of SAJO-2 in prior joint work from our groups through differential fluorination enhanced its antimicrobial potency; however, it also increased susceptibility to enzymatic digestion by β-trypsin. To address this limitation, the current research focuses on improving the overall efficacy of SAJO-2 through the incorporation of D-amino acids, beta backbone modifications, and a bulky pentafluorinated amino acid residue. All modified peptides exhibit resistance to enzymatic degradation, while antimicrobial activity was retained to differing degrees across organisms.
|
|
Scooped by
mhryu@live.com
June 5, 3:03 PM
|
Nanoparticles are now central to many drug and vaccine delivery strategies, but most require reformulation for each application. Bacteriophage‑derived nanoparticles offer a genetically encoded, structurally defined, and modular alternative. This review organizes recent advances along three tunable axes — scaffold, surface, and cargo — and highlights hybrid phage–polymer/lipid/inorganic constructs that expand stability, targeting, and loading. We survey applications from multivalent vaccines and oversized gene transfer to precision microbiome editing, and outline translational hurdles. Phage-derived products are approaching translation, with virus-like particle-based vaccines and CRISPR-enhanced antimicrobial phages in clinical trials. Finally, we preview emerging opportunities, including AI‑guided capsid and receptor‑binding protein design, cell‑free phage synthesis, and standardized ‘reference’ phage chassis that can be combined with traditional nanoparticles, positioning phage nanoparticles as reusable, plug-and-play nanomedicines.
|
|
Scooped by
mhryu@live.com
June 5, 2:31 PM
|
Cardioprotective effects of current therapies for mitigating ischaemia/reperfusion (I/R) injury have had limited success. The major challenge is to effectively control oxidative stress while preserving mitochondrial function in a timely manner. Hydrogen (H2) selectively reduces cytotoxic oxygen radicals, aiding in the regulation of physiological and pathological functions. However, the efficacy of H2 therapy is highly dependent on the amount and rate of H2 release, making it critically important to develop rapid, simple and efficient techniques for evolving therapeutic H2. Here we encapsulate H2-producing photosynthetic bacteria (PSB) in an injectable porcine dermal extracellular matrix (ECM) hydrogel to facilitate cardiac I/R injury repair. Upon light exposure, sustained and high H2 production from PSB hydrogel preserves mitochondrial homeostasis and essential functions. In a porcine model of cardiac I/R injury, PSB hydrogel treatment effectively mitigates myocardial damage and salvages jeopardized myocardium. We anticipate that this bacterial therapy for photosynthetic H2 production could provide an improved treatment for I/R-related diseases. Photosynthetic bacteria embedded in an injectable hydrogel produce ROS-scavenging species for mitigating myocardial damage and improving heart repair in a porcine model.
|
|
Scooped by
mhryu@live.com
June 5, 12:37 AM
|
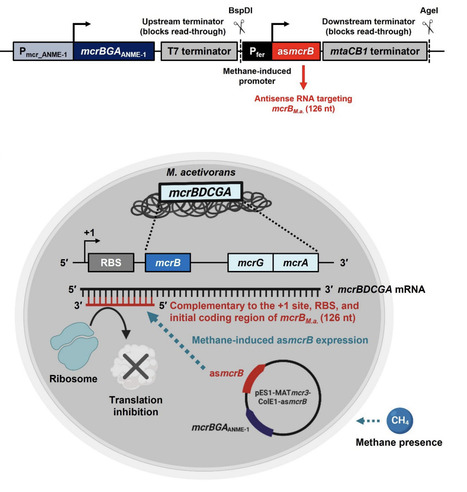
Utilizing methane and carbon dioxide before it can enter the upper atmosphere is beneficial for mitigating climate change as well as for producing valuable chemicals. Because anaerobic methanotrophic archaea (ANME) have not yet been cultured in isolation, we previously reversed methanogenesis by cloning the genes encoding methyl-coenzyme M reductase (Mcr) derived from Black Sea ANME-1 into the methanogen Methanosarcina acetivorans. The resulting engineered archaeal strain captures, rather than produces, methane and may be used to convert methane and carbon dioxide into electricity, acetate, L-lactate, and ethanol. However, the engineered M. acetivorans strain also contains a chromosomal locus encoding its native Mcr (McrM.a.), which produces methane from substrates such as methanol, whereas the heterologously expressed ANME-1 Mcr (McrANME-1) promotes methane oxidation. Therefore, we reasoned that McrM.a. may compete with McrANME-1-mediated reversal of methanogenesis. To enhance the reversal of methanogenesis, here we implemented an antisense RNA (asRNA) silencing approach to suppress McrM.a. during growth on methane while still allowing its expression during routine growth on methanol. We found that silencing McrM.a. during McrANME-1-mediated growth on methane increased ethanol and acetate production by more than an order of magnitude. These results were corroborated by both a more than 10-fold increase in methane utilization by McrANME-1 and a greater than 1,000-fold reduction in the McrM.a. mcrBGA transcript levels under methane-grown conditions. Therefore, asRNA-mediated silencing may be used to enhance methane capture by suppressing production of the host McrM.a. for biotechnological applications.
|


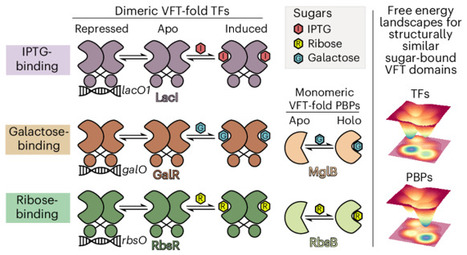




















2st, Using HX-MS they measured residue-level stability across apo, DNA-bound, and inducer-bound states, revealing that despite nearly identical structures across functional states, the energy distribution shifts dramatically between states. A helix at the dimer interface, H32, was consistently more stable in the DNA-bound state and less stable in the inducer-bound state.
redesigned the seven residues within H32 using ProteinMPNN, keeping the backbone fixed but changing side chain identities to tune intrinsic helix stability. This produced six LacI variants with IPTG sensitivities spanning four orders of magnitude, demonstrating that engineering the allosteric signal propagation pathway — rather than the ligand binding site itself — is a rational and predictable strategy for tuning transcription factor responses.