Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 3:25 PM
|
Since its discovery, the CRISPR-Cas system has ushered in a transformative era in biodetection, leveraging its simplicity and efficiency to enable Cas protein-based signaling systems for applications in early tumor screening, viral detection, and molecular logic circuits. However, the constrained compatibility of CRISPR/Cas-based signaling systems with diverse input types limits their versatility, primarily due to the restricted activation mechanisms of the Cas protein. Herein, we developed the Cas12a and Cas13a Integrated Targeting (CACIT) system, which harnesses DNA/RNA strand displacement reactions to integrate the enzymatic capabilities of Cas12a and Cas13a. This system supports simultaneous DNA and RNA inputs, offering exceptional programmability and cost-effectiveness. By employing strand displacement reactions, the CACIT system achieves synchronized activation of Cas12a and Cas13a. We have demonstrated that the CACIT system excels in single-nucleotide-variant (SNV) detection, viral RNA detection, machine learning-driven nucleic acid concentration response modeling, logic operations, and intracellular imaging. As a streamlined and versatile signaling platform, the CACIT system expands the scope of CRISPR/Cas activation strategies. With its inherent simplicity and compatibility, this system facilitates integration with diverse nanodevices. Further, this system provides a highly programmable, multifunctional computational module for molecular networks, heralding new possibilities for artificial signaling systems.
|
|
Scooped by
mhryu@live.com
Today, 3:06 PM
|
Adenosine triphosphate (ATP) hydrolysis is the main cellular source of energy used to drive biochemical reactions that are otherwise energetically unfavorable. The chemical energy stored in phosphoanhydride bonds is released upon hydrolysis of ATP to ADP and is used to drive mechanical work and conformational change. DNA replication is a canonical process in which the multi-enzyme replisome is thought to rely on ATP hydrolysis for its function. Here we show, through single-molecule visualization of DNA replication by the E. coli replisome, that the replicative DnaB helicase does not rely on hydrolysis of ATP in the context of the elongating replisome. Even in the presence of physiologically-relevant concentrations of ATP, dTTP is hydrolysed preferably. Finally, we show that the replicative helicases from S. cerevisiae, D. melanogaster, and Homo sapiens can also use dTTP to unwind DNA. Our observations suggest that replicative helicases across domains of life are ‘flex-fuel’ helicases. Here the authors show that replicative helicases from bacteria to humans can use dTTP instead of ATP for DNA unwinding, and that the E. coli replisome preferentially uses dTTP during DNA replication, challenging textbook models of replication energetics.
|
|
Scooped by
mhryu@live.com
Today, 1:08 PM
|
Bacterial plant pathogens cause hundreds of millions of dollars in annual crop losses in the United States alone, further stressing an already strained global food supply. Numerous bacteria in the phyllosphere use N-acyl homoserine lactone (AHL)-mediated quorum sensing (QS) systems to coordinate behaviors and shape microbial communities. Strategies that interfere with bacterial QS systems employ enzymes to degrade or transform AHLs in a process called quorum quenching (QQ). This interference results in altered microbial community structure and function. We previously isolated and engineered SsoPox, a highly stable AHL-degrading lactonase that interferes with QS. Here, we evaluated the effects of SsoPox-mediated QS disruption on the Zea mays phyllosphere in a Goss’s wilt infection model caused by Clavibacter nebraskensis. In this proof-of-concept work, we found that infection significantly altered the composition and structure of the leaf microbial community. SsoPox QQ lactonase treatment substantially reduced this shift, resulting in a leaf surface community resembling the uninfected controls. qPCR experiments revealed a small, yet significant reduction of pathogen abundance on plant leaves following enzymatic treatment. As a result, the formulated QQ lactonase spray reduced disease severity. This study demonstrates the critical role of microbial signaling in the phyllosphere and highlights the potential of QQ lactonases to control plant diseases.
|
|
Scooped by
mhryu@live.com
Today, 2:10 AM
|
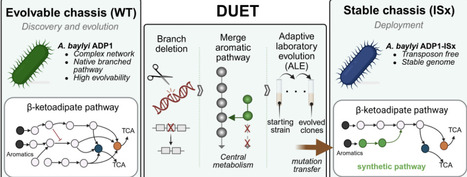
Genome streamlining and pathway refactoring are powerful strategies for constructing controllable microbial chassis for both fundamental studies and applications. While rational design benefits from reduced genetic complexity, adaptive laboratory evolution (ALE) thrives on metabolic redundancy, creating a mismatch between optimal hosts for design and evolution. Here, we introduce a dual chassis framework (DUET) in which rational pathway construction and adaptive evolution are first carried out in an evolution‑competent host, and the resulting optimized designs are subsequently transferred into a genetically stable chassis for deployment. Using the naturally evolvable bacterium Acinetobacter baylyi ADP1 and its genome-stabilized derivative (ISx), we applied this framework to the β-ketoadipate pathway, a central hub for aromatic compound catabolism. We first streamlined the native network by deleting individual pathway branches and then engineered a minimal synthetic route that merges protocatechuate and catechol metabolism. Subsequent ALE enabled efficient growth through this synthetic pathway, and reverse-engineering identified key adaptive mutations underlying functional recovery. Both the synthetic pathway and the mutations were transferred unchanged into ISx, where robust growth was maintained without further adaptation. These results demonstrate that DUET enables portable, host-independent deployment of rational metabolic streamlining combined with evolution, providing a generalizable strategy for building reduced yet robust microbial platforms.
|
|
Scooped by
mhryu@live.com
Today, 2:00 AM
|
Archaea have proven to be major players in biogeochemical cycles across diverse ecosystems, yet we still see an underrepresentation of archaeal genomes in the datasets used by popular computational biology tools. Here we present ArchaeaHQ, a quality-controlled, systematically curated reference database of 21,644 archaeal genomes compiled initially from 35,993 assemblies from all four archaeal kingdoms retrieved from NCBI: Methanobacteriati (Euryarchaeota), Thermoproteati (TACK), Nanobdellati (DPANN), and Promethearchaeati (Asgard). All genomes in the database passed standardized quality control, requiring ≥70% completeness and ≤10% contamination. A total of 44.2% of genomes in ArchaeaHQ achieved ≥90% completeness, while 93.1% exhibited ≤5% contamination. ArchaeaHQ comprises 16,199 metagenome-assembled genomes (MAGs; 74.8%) and 5,445 isolate genomes (25.2%). Approximately 75% of MAGs are assigned to 17 ecologically meaningful categories based on sampling origin, and around 65% of genomes include geographic metadata. ArchaeaHQ is available at https://doi.org/10.6084/m9.figshare.32266599 and provides an analysis-ready reference set for metagenomic classification, biogeochemical and ecological studies, comparative genomics, and development of archaeal-specific bioinformatic tools.
|
|
Scooped by
mhryu@live.com
Today, 1:48 AM
|
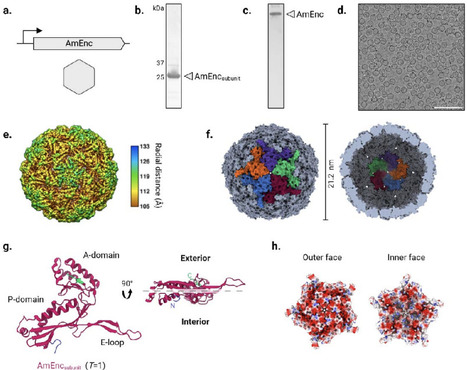
Encapsulins are prokaryotic self–assembling protein nanocages with promise as nanovaccine scaffolds. Their utility as modular platforms require tolerance to surface engineering, high-yield soluble production, formulation stability, and controlled antigen (co–)display. Herein, a previously uncharacterized encapsulin from Alkaliphilus metalliredigens is engineered into a SpyCatcher–decorated nanoscaffold (Am–S) that enables controlled surface display of SpyTagged antigens. Cryo-EM confirms that the native encapsulin forms a T = 1 icosahedral nanocage, and that C-terminal SpyCatcher fusion yields Am–S without compromising nanocage assembly, symmetry, or structural integrity. Notably, Am–S exhibits high-yield soluble production in Escherichia coli, remains monodisperse after freeze–thaw and extended storage, and supports efficient SpyTagged peptide conjugation for single– and multi–antigen display. As a proof–of–concept, Am-S is functionalized with Alzheimers disease-associated beta–amyloid and/or hyperphosphorylated tau epitopes to generate single–target nanocages displaying either antigen and dual–target mosaic nanocages co–displaying both. In mice, Am–S antigen display enhances antigen–specific IgG responses relative to free antigens and induces predominantly IgG1–biased humoral immunity. Mosaic nanocages elicit antibodies against both targets, with immune sera selectively recognizing amyloid–beta and phosphorylated tau-associated pathology in ex vivo brain sections from Alzheimers disease mouse models. These findings position Am–S as a manufacturable scaffold for developing multi–targeting nanovaccines against complex diseases.
|
|
Scooped by
mhryu@live.com
Today, 1:40 AM
|
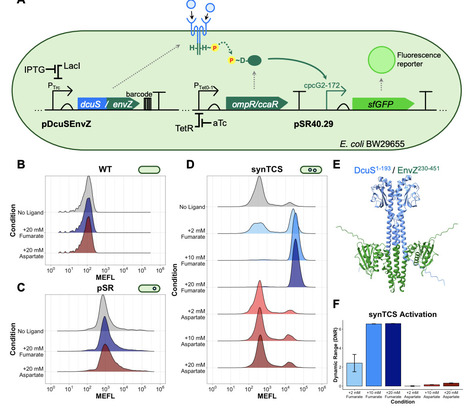
While bacterial sensor histidine kinases (SHKs) are widespread as natural molecular biosensors, tools for high-throughput characterization of SHK signaling phenotypes are limited, hindering wide scale implementation of bacterial-based sensing. Here, we developed a synthetic two-component signaling system that reports chimeric SHK signaling via a standardized fluorescence readout. With this synthetic system, we screened a library of chimeric DcuS/EnvZ SHKs to characterize sequence-function relationships within in the DcuS sensory and transmembrane domains. We quantified the effects of 1,173 mutations on signaling outputs in the presence of fumarate, a native DcuS ligand, as well as aspartate for which DcuS has minimal affinity for. We identified eleven positions across the DcuS domains which significantly alter aspartate responsiveness and selectivity and further observed a role for cytoplasmic N-terminal residues in determining signaling outputs. In future studies, this framework will expedite design of biosensors for novel ligands by enabling high-throughput screening of mutagenized libraries of natural SHKs.
|
|
Scooped by
mhryu@live.com
Today, 1:33 AM
|
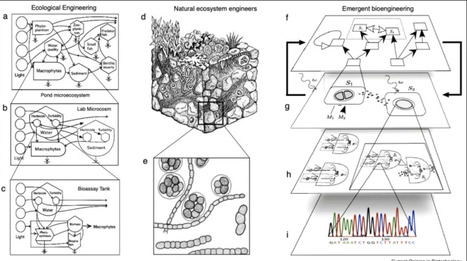
The biosphere is undergoing an unprecedented transformation driven by global warming, habitat loss, and resource depletion, threatening biodiversity through widespread species extinctions and population declines. Although conservation and restoration remain essential, the risk of irreversible tipping points demands new strategies. Synthetic biology offers one such approach: engineering existing ecosystems by modifying functional traits of resident communities to enhance resilience and prevent abrupt shifts. Despite and because of public concern, advances in biosafety and control have been achieved, mainly on a cellular scale. However, after decades of bioremediation efforts, a central question emerges: not only can interventions be perfectly controlled, but also whether they can persist and sustain ecological function. Meeting this challenge requires a paradigm shift in design philosophy, from classical to emergent engineering, embracing adaptation, feedback, and multiscale complexity as the foundation of ecosystem design.
|
|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
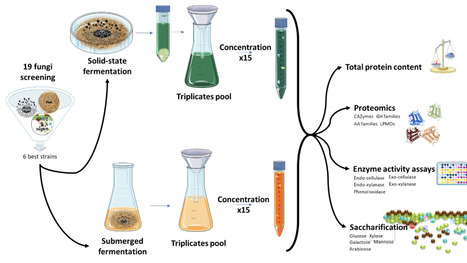
Lignocellulosic biomass represents a promising renewable feedstock for sustainable biorefinery applications, yet efficient enzymatic saccharification remains challenging due to the recalcitrant structure of plant cell walls. This study presents a comprehensive comparative analysis of enzymatic activities, saccharification performance, and secretome composition of six fungal species cultivated under solid-state fermentation (SSF) and submerged fermentation (SmF) conditions using untreated flax shives as substrate. While SmF yielded approximately 4-fold higher total protein concentrations (0.38 ± 0.13 g.L-1 vs. 0.08 ± 0.02 g.L-1), SSF-derived enzymes demonstrated superior specific enzymatic activities, particularly for endo-xylanase and endo-cellulase, resulting in more efficient biomass saccharification. Proteomics analysis revealed distinct secretome profiles between fermentation modes, with SSF showing higher proportions of polysaccharide metabolism proteins (71.0%) compared to SmF (49.3%), while SmF exhibited greater enzyme diversity including more lytic polysaccharide monooxygenases (LPMOs) and auxiliary activity enzymes. Trichoderma species consistently demonstrated the highest saccharification efficiency, with glucose yields reaching 2.37 mM under SSF conditions. A Scheffe simplex-lattice mixture design comprising 65 enzyme cocktail combinations revealed significant synergistic interactions between several cocktails, with the binary mixture of Trichoderma 2SA21 and P. chrysogenum achieving 54% synergy - in terms of higher sugar release above expectations - and the highest total monosaccharide release (1.80 mM). These findings provide practical guidance for developing cost-effective enzyme cocktails for lignocellulosic biorefinery applications, emphasizing the importance of fermentation mode selection and strategic strain combination over enzyme supplementation complexity. The methodology established here, combining systematic screening, comparative proteomics, and statistical mixture design, offers a robust framework for optimizing fungal enzyme systems across diverse biomass substrates.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
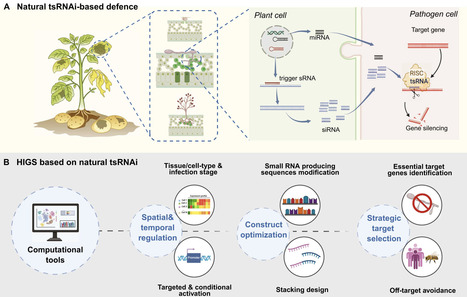
Host-induced gene silencing (HIGS) is a crop protection strategy that exploits RNA interference (RNAi) to silence targeted genes in invading pathogens or pests and reduce disease. Despite some successful examples of HIGS in laboratory settings, its translation into commercial agriculture has been limited. Recent discoveries demonstrating that plants deploy specific endogenous small RNAs (sRNAs) to regulate gene expression in fungi and oomycetes have broadened our understanding of natural trans-species RNAi (natural-tsRNAi) and provided a framework for improving applications of sRNA-based defense. In this review, we summarize HIGS studies published between 2021 and 2025 with a meta-analysis, highlighting their potential and limitations. We then discuss recent advances in natural tsRNAi with an emphasis on the secondary small interfering RNA pathway as a native immune response. Finally, we provide our opinion on how insights from natural tsRNAi inform design principles for sRNA-based immunity as a promising source of engineering durable resistance traits.
|
|
Scooped by
mhryu@live.com
June 3, 5:15 PM
|
The infectious diseases remain a primary cause of morbidity and mortality on a global level, a crisis exacerbated by the rapid emergence of antimicrobial resistance (AMR). The ineffectiveness of the old antibiotics and the stagnation of the development of new medications have triggered an emergency in the search for new methods of treatment. This review examines the opportunity of nanotechnology, which is the so-called nanobiotics, as a game-changing technology in combating multidrug-resistant (MDR) bacteria by searching various databases. Nano-particles (NPs) are unique physicochemicals, i.e., high surface-to-volume ratio and multi-valent properties that enable them to overcome traditional resistance, e.g., efflux pumps, target modifications. The paper discusses the important antibacterial processes, which can involve the induction of oxidative stress by the production of reactive oxygen species (ROS), the physical destabilization of the bacterial cell envelope, or the targeting of intracellular macromolecules, such as DNA and proteins. Moreover, it emphasizes the synergistic nature of the interactions of NPs with traditional antibiotics to improve drug delivery and efficacy and decrease host toxicity. Although a wide range of nanomaterials, such as silver, gold, metal oxides, and carbon-based structures, promise a lot, a lot of questions have been raised on their compatibility in the long term, their impact on the environment, and their legality. Nanotechnology, coupled with precision medicine and CRISPR-based systems, is a promising future in dealing with infectious diseases. Finally, nanomedicine is a complex solution to address the shortcomings of existing treatments and achieve a future of world health.
|
|
Scooped by
mhryu@live.com
June 3, 5:13 PM
|
Liquid–liquid phase separation (LLPS) plays a central role in cellular regulation, with its dysregulation linked to numerous diseases. LLPS is also increasingly implicated in various biological contexts, such as virus replication. These findings have driven the development of numerous computational predictors to screen and identify phase-separating proteins from sequence and/or structural models. Despite the need for these tools, their performance across diverse biological contexts remains incompletely understood, complicating tool selection and result interpretation. We performed a systematic comparative analysis of 9 LLPS prediction algorithms using multiple curated datasets comprising both LLPS-positive (LLPS+) and LLPS-negative (LLPS−) proteins. The datasets span multiple biologically relevant scenarios, including intrinsically disordered proteins, folded proteins, proteins with LLPS-abolishing variants, benchmark datasets, and viral proteins. We observed substantial variability in predictive performance across algorithms when assessing proteins of different structural classes such as LLPS+ and LLPS− folded proteins, LLPS-abolishing mutations, and viral proteins. These results demonstrate that LLPS predictor performance is strongly context dependent, leading to different predictors being optimal for different biological questions. For overall protein assessment, DeePhase and MolPhase provided the most consistently accurate predictions, being the least impacted by structural bias. For assessing the impact of small mutations on LLPS propensity, PSPHunter, a built-for-purpose algorithm, reliably predicts mutation impacts, with structure-informed algorithms PSPire and PICNIC also providing strong insight. Across all evaluated datasets, the findings highlight the need for well-benchmarked training and testing data that encompass a broad and diverse range of protein classes.
|
|
Scooped by
mhryu@live.com
June 3, 5:08 PM
|
Bacterial-mediated cancer therapy shows therapeutic potential yet remains constrained by bioavailability and biosafety challenges. We introduce a scalable, modular biosurface-engineering platform for functionalizing wild-type magnetotactic bacteria (Magnetospirillum magneticum, AMB-1), enabling the integration of diverse materials, including small molecules, polymers, nanoparticles, and metal–organic frameworks. As a proof-of-concept, AMB-1@Fe3+-PDA (poly dopamine) exemplifies a bacteria-mediated therapeutic strategy that merges tumor-targeted delivery, immunosuppressive tumor microenvironment (TME) reprogramming, and innate immune activation with photothermal-chemodynamic therapy. The Fe3+-PDA coating counteracts deep-tissue immunosuppression and primes adaptive immunity, while AMB-1 enhances penetration into resistant TME niches. These mechanisms synergistically suppress aggressive 4T1 breast tumor progression, metastasis, and recurrence while establishing immunological memory. In murine models, two treatment cycles prolonged median survival from 45 to 67 days and elicited systemic antitumor immunity, including abscopal effects targeting untreated distant tumors. This platform establishes a bridge between materials science and synthetic biology, providing a generalizable blueprint for advancing the clinical translation of engineered bacterial therapies.
|
|
|
Scooped by
mhryu@live.com
Today, 3:10 PM
|
Protein-based biosensors offer unique advantages over conventional analytical methods by enabling real-time detection of target analytes with minimal sample preparation. However, efficiently coupling molecular recognition to a reliable output signal remains a key challenge in biosensor design. Here, we present a plug-and-play strategy using the de novo switch platform, LOCKR, which enables direct transduction of a binding event into a defined signal output. The LOCKR architecture supports modular reconfiguration: the recognition domain can be swapped to detect a desired analyte, while the reporter module can be interchanged to tune the output format. We expand the range of LOCKR-compatible readouts beyond split luciferase to include ratiometric Förster-type resonance energy transfer and β-lactamase-based colorimetry. By integrating computationally designed high-affinity binders as interchangeable recognition elements, we demonstrate sensitive detection of glucagon, neuropeptide Y, and peptide YY with limits of detection in the picomolar range. Together with the expanding landscape of de novo designed binders and reporters, the LOCKR platform bridges the gap between molecular recognition and signal generation, enabling versatile biosensor development tailored for user-defined applications.
|
|
Scooped by
mhryu@live.com
Today, 1:21 PM
|
Extracellular vesicles are cell-derived secretions that mediate tissue homeostasis and intercellular communication through their diverse cargos, including proteins. Distinct extracellular vesicle biogenesis pathways suggest specific association and co-enrichment of proteins sharing a biogenesis pathway, and non-association and co-depletion of proteins segregated into distinct pathways. Yet these associations elude conventional protein expression or co-expression measurements. Here, we propose and define pairwise protein co-enrichment relative to its overall expression to quantify whether a given protein is co-enriched or co-depleted with another protein. We measure co-enrichment, and differential co-enrichment between a stimulus and a reference condition, of up to 240 protein pairs in extracellular vesicles using antibody microarrays. We validate co-enrichment by modulating well-known extracellular vesicle biogenesis pathways, and find that differential co-enrichment measures expected changes between perturbed and reference conditions. Co-enrichment and differential co-enrichment in three model cell lines and parental and organotropic breast cancer progeny cell lines reveal both preserved and variable co-enrichment that may warrant further studies. Collectively, our result suggest that co-enrichment reflects cell physiology and extracellular biogenesis, is readily measurable, and could serve as quality control in extracellular vesicle biomanufacturing. Distinct extracellular vesicle biogenesis pathways suggest specific co-enrichment of proteins sharing a biogenesis pathway. Here, the authors design an approach to examine this co-enrichment and show that it reflects vesicle biogenesis.
|
|
Scooped by
mhryu@live.com
Today, 1:02 PM
|
Arsenic (As), a toxic element widespread in paddy soils worldwide, is mobilized by microbial processes, posing risks to environmental quality, food safety and human health. The rice rhizosphere is a dynamic environment with day–night cycles in chemical conditions and microbial activity. However, how arsenic itself changes over these daily cycles remains unexplored. Here we conduct metatranscriptomic and biogeochemical analyses in a greenhouse study to investigate the diurnal rhythms of As dynamics in the rice rhizosphere. We observed consistent diel fluctuations in arsenite (As(III)) concentrations, increasing from 1.8 to 2.9 mg l‒1 at night, alongside a 24.9% rise in ferrous iron (Fe(II)). Redox potential decreased to ~100 mV at night, promoting As/Fe reduction. Transcriptional activity of key functional genera involved in dissimilatory As/Fe reduction (for example, Geomobilimonas and Geobacter) increased at night, reflected in higher transcript abundances of reduction genes (arrA, omcS, omcZ and mtrC), without corresponding changes in relative abundance. These patterns were confirmed in a field study. Under constant darkness, these diel patterns disappeared. Together, these findings suggest managing rice cultivation to align with natural daily cycles may reduce contamination risks and optimize nutrient management. Diurnal cycles in microbial reducer activity reveal synchronized mobilization of arsenic and iron in the rice rhizosphere, as shown by metatranscriptomic and biogeochemical analyses.
|
|
Scooped by
mhryu@live.com
Today, 2:04 AM
|
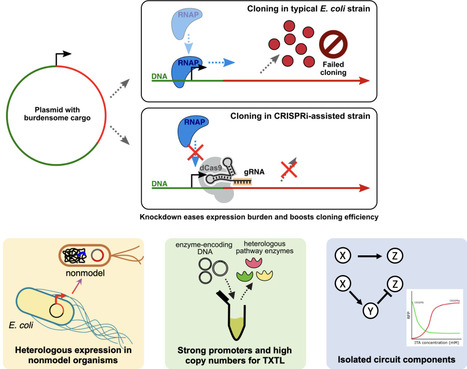
Genetic constructs meant for metabolic engineering in nonmodel microbes often use similar genetic parts to those familiar to E. coli work. The typical workflow is to clone these parts into plasmids in E. coli before they are transferred to the nonmodel host or its genome. In many cases, the metabolic burden of these constructs is stronger in the E. coli cloning phase of the workflow than in the eventual host, possibly resulting in mutation or other failure during cloning. Here, we apply generic knockdown of a range of popular expression systems, using CRISPR interference, by targeting guide RNAs to either promoters or RBSs that are commonly used in metabolic engineering. Generic targeting of a constitutive promoter series, combined with genome integration of CRISPR components, allows the use of only one or a few specific cloning strains to achieve strong knockdown of a wide range of constructs. Further, we present a recombinase-based workflow for easily adding guide RNAs with custom targets, so that users can knock down any desired promoter or ORF. Together, this group of strains comprises easy-to-use cloning strains meant for increasing success rates of difficult or burdensome cloning reactions, ultimately allowing more ambitious genetic constructs to reach their intended context.
|
|
Scooped by
mhryu@live.com
Today, 1:56 AM
|
Understanding how sequence context influences the likelihood of mutation at a given genomic locus is critical for deciphering evolutionary processes. It is well established that the immediately adjacent bases influence mutation rates, but the role of more distal bases remains poorly understood. Here, we analyze over 100,000 mutations from 32 E. coli mutation accumulation experiments, encompassing strains with varying DNA proofreading and mismatch repair capabilities. By quantifying the frequency of each nucleotide up to 6 bp around mutation sites, we reveal complex mutational biases that extend beyond the immediately adjacent bases and are unique to each type of base pair substitution. Furthermore, which sequence contexts contribute most to mutational bias depends on what repair mechanisms are active and which strand serves as the leading versus lagging template during replication. Mononucleotide runs are prominent mutational hotspots–we systematically characterize which types of runs are the most mutagenic, including a previously undescribed hotspot for G:C→C:G transversions that can increase their frequency by up to four orders of magnitude. Remarkably, extending our analysis to 1,000 bp from mutation sites reveals that sequence context can influence mutational bias over unexpectedly long distances. These findings expose the intricate interactions between extended sequence context and DNA repair systems that shape spontaneous mutagenesis, and shed light on the mechanistic origins and evolutionary consequences of mutational signatures.
|
|
Scooped by
mhryu@live.com
Today, 1:44 AM
|
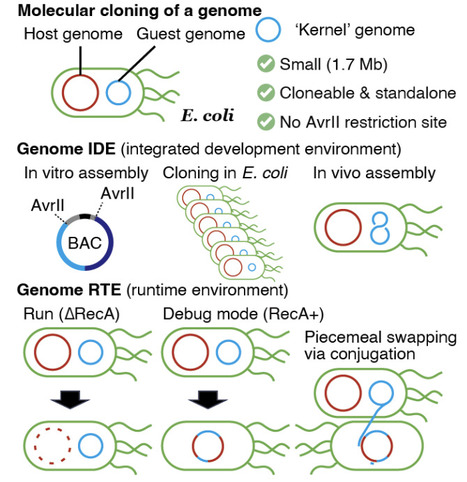
Genome synthesis is a major limitation in generative biology. Here, the half-sized genome of E. coli was constructed by fleshing out an imperfect minimal genome through genome-scale debugging process. Our platform consists of integrated development environment (IDE) and runtime environment (RTE). The genome IDE supported the cell-free assembly of 200-300 kb plasmids and their in vivo fusion into a single 1.7 Mb plasmid. This imperfect genome was stably maintained in E. coli as a guest genome. The RTE relies on the restriction enzyme-mediated self-digestion of the host genome in the presence and absence of the RecA recombinase. The guest genome was tested, debugged, and partially replaced by the host genome to establish E. coli controlled by a 2.3-Mb genome. This is less than half in size of the wildtype and the smallest ever reported. Enfleshing a guest genome will facilitate genome printing that transforms AI-designed genomes into physical ones.
|
|
Scooped by
mhryu@live.com
Today, 1:36 AM
|
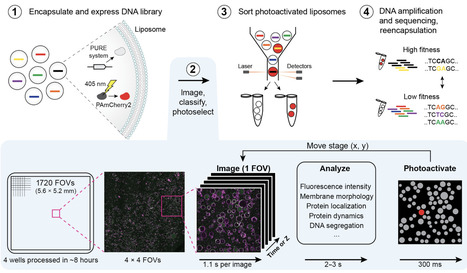
Understanding the relationships between genotype and phenotype is key to many areas of biological research and to the development of synthetic cells. We describe an image-based screening and sorting workflow that explores the phenotypes of gene-expressing vesicles within nonclonal populations and selects the desired variants. Using automated confocal microscopy and real-time, neural network–assisted image analysis, we demonstrate that liposomes can be selected for fluorescence intensity, protein localization, membrane morphology, and dynamic behaviors, and their phenotype can be linked to genetic content. This approach could substantially accelerate the evolution of cellular functions in a minimal synthetic context.
|
|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
Bacteriophages are crucial components of the human microbiome and hold promise as therapeutic agents. Yet, their physical interactions with mammalian cells remain poorly understood. Here, we developed a high-throughput platform to identify phages that adhere to epithelial layers and the proteins that mediate this interaction. The identified phages encode immunoglobulin (Ig)-like domain-containing proteins that, when displayed on a non-adherent phage, confer epithelial binding and internalization in vitro, and increased phage retention in the mouse gut in vivo. Phages encoding these adhesins are among the most abundant and prevalent human gut phages, including crAss-like phages and myoviruses closely related to the recently proposed Flandersviridae family. Domain sequence variation alters epithelial interaction profiles, and internalized phages traffic to the endoplasmic reticulum through the Golgi apparatus, suggesting access to non-degradative internalization pathways. These findings reveal widespread phage-human interactions in the human viral community, with potential impacts on health and implications for next-generation phage therapeutics. Here, the authors reveal that abundant gut bacteriophages use adhesive proteins to bind, enter, and traffic inside human epithelial cells, reshaping how phages are understood in the microbiome and opening paths for future phage-based therapies.
|
|
Scooped by
mhryu@live.com
Today, 1:03 AM
|
For decades, natural product biosynthetic enzymes have challenged conventional logic and expanded the repertoire of biological catalysis. Understanding these enzymes that catalyze such diverse and powerful reactions is critical for understanding biological pathways, leveraging biomimetic or biocatalytic chemistry, and discovering novel natural products. Uncovering these enzymes is a challenging and historied pursuit, where the landscape has shifted greatly with advances in bioinformatics and unprecedented access to large datasets. These new omics technologies have unlocked new strategies, complementing older strategies, for the prioritization of candidate enzymes for characterization. This Highlight examines several established and emerging strategies for uncovering novel enzymatic chemistry in the context of natural product biosynthesis, focusing on representative examples that define current capabilities and outline key challenges.
|
|
Scooped by
mhryu@live.com
Today, 12:19 AM
|
Enzyme engineering and metabolic engineering drive innovation in energy biotechnology. In recent years, artificial intelligence (AI) has supported successful applications in designing effective enzymes and productive microbial cell factories. This review summarizes recent advances in enzyme redesign using protein language models, de novo enzyme design with generative models, and AI tools for engineering metabolism and related cellular phenotypes. Across these areas, AI models are shifting from single modality inputs to integrated representations of protein function, metabolic pathways, and cell states. We emphasize that unifying the diverse data representations across scales will be necessary for advancements in energy biotechnology.
|
|
Scooped by
mhryu@live.com
June 3, 5:14 PM
|
Accurate prediction of enzymes' optimal catalytic temperature (Topt) is crucial in biotechnology, as enzymes with extreme Topt values are highly desirable for reactions at extreme temperatures and for their general stability. However, experimental determination of Topt is costly, labor-intensive, and time-consuming. Meanwhile, existing computational methods suffer from small and imbalanced datasets, suboptimal predictions at extreme temperatures, and insufficient validation. In this study, we address these challenges by expanding the Topt dataset and validating on an independent test set based on sequence similarity. We further tackle these limitations by comparing multiple resampling techniques to improve predictions at extremes and by considering diverse protein representations and multiple machine learning architectures. Overall, the best performing models reached R2 approximately 0.64 with MAE approximately 7-8 degrees C, while extreme resampling improved tail performance, reducing tail MAE by up to approximately 1.8 degrees C. Notably, our models show improved performance over state-of-the-art prediction models. We also demonstrate that accurate prediction of Topt is achievable even in the absence of organism growth temperature (OGT). Our Topt prediction models are made freely available as AdventML on GitHub.
|
|
Scooped by
mhryu@live.com
June 3, 5:10 PM
|
Artificial intelligence (AI) has transformed protein engineering by leveraging deep learning, protein language models, and knowledge graphs to decode relationships between sequence, structure, and function. Models like AlphaFold2 achieve near-experimental accuracy in structure prediction, while transformer-based language models facilitate de novo sequence design under functional constraints. AI enhances therapeutic protein engineering, enzyme catalysis, and synthetic biology, accelerating the transition from in silico design to experimental validation. These advances accelerate experimental validation across healthcare and industrial biotechnology. Despite algorithmic successes, challenges remain in model interpretability, training data biases, and experimental validation rates. This review examines the computational methodologies shaping protein design, benchmarking metrics, and the integration of machine learning with experimental pipelines.
|