 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:29 AM
|
Compact RNA-guided nucleases such as IscB represent an attractive foundation for next-generation genome editors, yet their application in mammalian cells has been constrained by suboptimal activity. Here, instead of re-engineering enzymes, we establish a mutant-initiated, structure-guided optimization strategy to generate second-generation high-activity IscB editors. Using AlphaFold3 to model the engineered IscB*-ωRNA–DNA complex, we reveal remodeling of the nucleic-acid-binding interface induced by activity-enhancing substitutions. Guided by this predicted structure, we perform a focused mutational scan and identify V367 as an activity hotspot. Saturation mutagenesis at this position yields a single substitution, V367Y (IscB*-Act), which increases mean editing efficiency by 34% and achieves up to 2.1-fold improvement across endogenous targets in mammalian cells. Importantly, the V367Y substitution is transferable to an IscB-based adenine base editor, elevating A-to-G conversion by 68% on average and up to 4.46-fold at individual loci without altering the intrinsic editing window. Targeted off-target profiling at loci suggests that V367Y does not substantially increase off-target indels or A-to-G conversion. Together, our work demonstrates a practical framework for second-generation refinement of compact genome editors, bridging deep-learning-enabled structural prediction with interpretable protein engineering, and expands the functional potential of miniature IscB systems for both nuclease and base editing applications.
|
|
Scooped by
mhryu@live.com
Today, 1:18 AM
|
Polyploidy, also known as whole genome duplication, is a major evolutionary force in plants, driving diversification and the generation of novel phenotypic variation, including superior abiotic and biotic stress tolerance. The enhanced stress resilience observed in certain polyploids is hypothesized to arise from dynamic epigenetic and genetic changes, including variations in gene content and cis-regulatory elements (CREs), that emerge following polyploidization. These changes directly impact various regulatory, signaling, and metabolic pathways associated with stress response and adaptation. Within polyploid populations, processes like gene duplications, fractionation, and homoeologous exchanges actively shape novel gene content variation, while, simultaneously, alterations in CREs (DNA sequences controlling gene expression) lead to diverse regulatory patterns. This dynamic interplay between changes in gene content and regulation further contributes to expanded phenotypic variation, including enhanced stress resilience. We discuss how advanced genomic and epigenomic techniques, such as pangenomics and single-cell assay for transposase-accessible chromatin with sequencing, are used to uncover these variations and outline new bioinformatic approaches to reveal the underlying genetics of stress resilience and adaptation. Finally, we summarize what remains poorly understood to guide future research, with the goal of unlocking the full potential for enhancing resilience in polyploid crops.
|
|
Scooped by
mhryu@live.com
Today, 1:02 AM
|
Biological nitrogen fixation in some filamentous cyanobacteria occurs in heterocysts, yet the subcellular organization of nitrogenase in these cells remains poorly defined. Using fluorescence microscopy of living Anabaena sp. PCC 7120, we found that a NifH-GFP fusion forms discrete puncta restricted to mature heterocysts, whereas constitutively expressed GFP in vegetative cells and a nifB-linked GFP reporter remained diffuse. To contextualize this phenotype, we built a homology-aware cyanobacterial condensate-prioritization framework across 31,028 proteins from seven proteomes, integrating ESM-2 embeddings, 27 biophysical features, and 44 curated condensate-associated positives. Because direct positives remain limited, we present the model as a ranking resource rather than a calibrated classifier. Under a deliberately conservative nitrogenase-withheld analysis that removed nitrogenase-family labels and homologous clusters from the training data, NifH retained a median rank in the highest-scoring 4.8% of cyanobacterial proteins. catGRANULE 2.0 also assigned high scores to nitrogenase iron proteins, while having only modest rank concordance to the whole atlas. Together, these data identify heterocyst-restricted NifH-GFP puncta as evidence of sub-cellular spatial organization and provide a curated resource for prioritizing cyanobacterial condensate candidates, some of which are also important for nitrogen fixation.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
Tandem gene duplication drives antibiotic resistance, metabolic adaptation, and gene-family expansion in bacteria, but no tool detects them in reference genomes, discovers their junctions in isolate sequencing, and quantifies the junctions in population samples. Existing callers (e.g. breseq) detect duplications without classifying formation mechanisms and often fail to quantify the duplication. Tandem has 3 modules. Module 1 detects reference-genome duplications by NUCmer self-alignment and classifies each by homologous-recombination signature and the junction microhomology length. Module 2 confirms junctions in whole-genome sequencing at user-nominated coordinates after user inspecting the coverage plot. Module 3 quantifies known junction in population sequencing using the novel Junction Read Ratio (JRR). On 280 artificial population tests across seven bacterial species, Tandem achieves 100% recall and 4.3% mean absolute error. Applied to experimentally evolved Pseudomonas fluorescens SBW25 populations, Tandem resolves multiple co-segregating duplication fragments.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
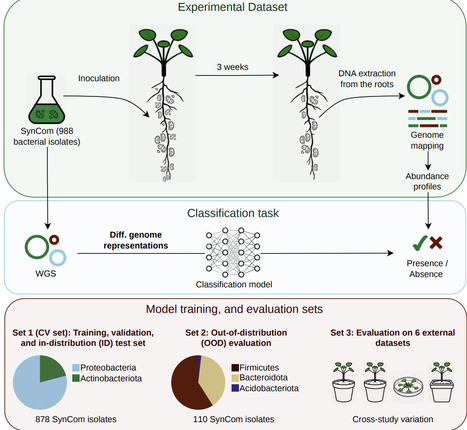
Root competence, the ability of soil bacteria to establish and grow on plant roots, is a key ecological trait influencing plant nutrition, growth, and health. However, identifying genomic determinants of root competence across bacteria remains challenging, in part because model generalizability depends strongly on how genomes are represented. Traditional approaches based on curated annotations are incomplete and biased toward well-characterised organisms and functions, limiting generalization. Sequence-similarity clustering improves coverage but yields high-dimensional features relative to dataset size, hindering training. Foundation models offer an alternative by learning compact representations without relying on prior annotation. Here, we compared pretrained genome representations from protein and DNA foundation models (ESM-2, Bacformer, DNABERT-S) with annotation- and clustering-based features (KEGG orthology, OrthoFinder protein families) for predicting root competence using synthetic microbial community data from Arabidopsis thaliana and assessed generalisability across bacteria. When training and test sets contained taxonomically related bacteria, most approaches performed similarly. However, when test bacteria belonged to phyla entirely absent from training, reflecting high evolutionary separation across all levels of bacterial classification, only pretrained protein representations retained predictive performance. Bacformer-derived representations, which incorporate genomic context, supported the strongest generalisation, suggesting that conserved genomic organisation contributes to predicting root competence. Feature attribution quantifying protein contributions to model decisions linked root competence to TonB/SusD-dependent receptors, small-molecule transporters, and unannotated proteins with conserved regulatory motifs and homology to carbon starvation-response loci. Protein foundation models support generalisation across evolutionarily distant bacteria and identify genomic determinants of root competence, including unannotated proteins.
|
|
Scooped by
mhryu@live.com
May 26, 7:29 PM
|
Building a living cell from scratch requires overcoming a bottleneck that has remained unresolved despite decades of progress: orchestrating the spatiotemporal integration of core functional modules. To tackle this barrier, the SynCell Asia Initiative outlines a strategy for developing core functional modules followed by their systems-level integration through the establishment of a centralized, artificial intelligence (AI)-driven biofoundry.
|
|
Scooped by
mhryu@live.com
May 26, 7:08 PM
|
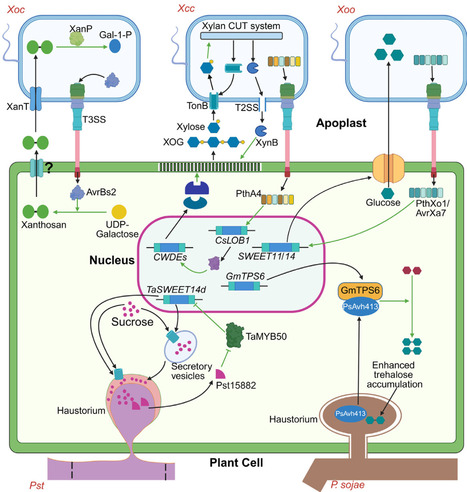
Besides suppressing immunity, pathogen effectors hijack host biosynthetic pathways, sugar transporters, enzymes, and transcriptional regulators for nutritional gain. In Xanthomonas, AvrBs2 drives de novo nutrient synthesis from a host metabolite, while PthA4 hijacks fruit ripening to release sugars. These findings pave the way for ‘anti-nutrition’ approaches for durable crop resistance.
|
|
Scooped by
mhryu@live.com
May 26, 6:03 PM
|
To meet the needs of a growing human population, agricultural management practices have undergone substantial intensification, specialization and industrialization. This has contributed to biotic homogenization and a loss of diversity in microbial communities within agricultural systems. In this Perspective, we summarize recent studies that report microbial homogenization due to agricultural intensification. We propose a definition of microbial homogenization and explore how intensive agricultural practices can cause taxonomic, physiological, genetic and functional homogenization of microbial communities. Our analysis indicates that globally the diversity of rare taxa is lower in intensively managed agricultural lands compared with less-intensive lands and that agricultural intensification suppresses beneficial microorganisms and promotes pathogenic taxa. We identify microbial taxa that are sensitive to intensification and discuss how the disproportionate impact on rare microbiota can threaten agro-ecosystem functions and food security. Finally, we outline key challenges and suggest areas that require further research. In this Perspective, Banerjee, Dasgupta and van der Heijden discuss how intensive agricultural practices can lead to the taxonomic, physiological, genetic and functional homogenization of microbial communities and disproportionately affect rare microbiota.
|
|
Scooped by
mhryu@live.com
May 26, 5:49 PM
|
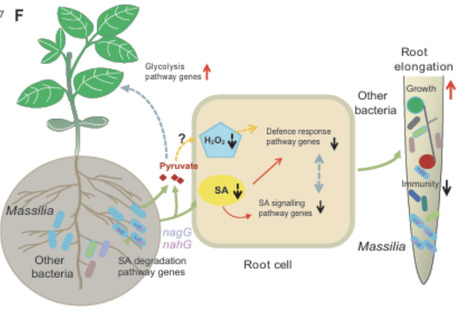
Rhizosphere microbiota play an important role in maintaining plant root growth. However, the physiological and molecular basis of microbial regulation of root traits remains poorly understood. Here, we report that Massilia efficiently colonizes roots and promotes root elongation. In particular, the M117 strain of Massilia significantly inhibits salicylic acid (SA)-related immune signalling, which is required for M117-mediated root elongation. M117 can directly degrade SA, thereby reducing SA levels in the roots. Integrated omics reveal the presence of multiple SA hydrolytic pathways in M117. Among them, the NagGHAaAb pathway is strongly induced by SA. This pathway regulates root growth and is nonrandomly distributed across Massilia species. Finally, we show that M117 colonization enriches specific bacterial taxa within roots. Our findings reveal a specific pathway employed by rhizosphere bacteria to colonize roots and promote their growth and highlight a useful microbial strategy and information for balancing host immunity and growth.
|
|
Scooped by
mhryu@live.com
May 26, 5:36 PM
|
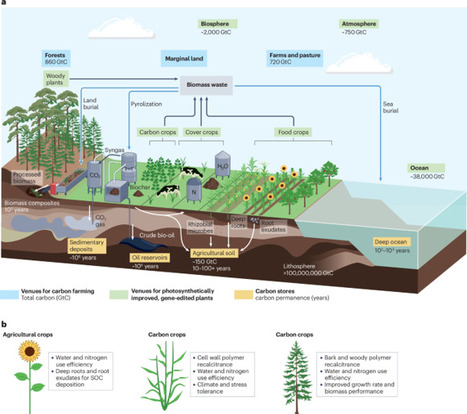
Anthropogenic carbon emissions have destabilized Earth’s carbon cycle, triggering cascading effects on climate and biodiversity. Plant-based carbon dioxide removal (CDR) presents a scalable, economically viable path to atmospheric carbon sequestration through soil carbon deposition, dedicated biomass cultivation and strategic agroforestry. Although photosynthesis drives terrestrial carbon capture, effective CDR strategies demand genetic optimization of carbon assimilation, retention and storage. The regulatory landscape is restrictive towards transgenic crops yet permissive of genome editing, creating a window for intervention. Advances in CRISPR-based editing, computational plant trait prediction and delivery systems for gene-editing tools in planta enable precision engineering of plant phenotypes to increase photosynthetic efficiency and carbon sequestration capacity. In this Review, we map the molecular and physiological innovations required to realize plant-based CDR at climate-relevant scales. Beyond optimizing carbon capture itself, we examine strategies to engineer enhanced biomass accumulation, improve nitrogen and water use efficiency, and stabilize carbon storage in plant and soil systems. We further assess the opportunities, implementation challenges and the potential of deploying genome-edited crops as a cornerstone of global carbon management. Gene editing to enhance photosynthesis in crop plants offers a strategy to boost plant carbon capture and mitigate climate change. This Review explores the agronomic, molecular and technical challenges in engineering photosynthesis for carbon sequestration as well as the genetic tools and editing technologies that can improve plant productivity and carbon storage.
|
|
Scooped by
mhryu@live.com
May 26, 5:25 PM
|
Rhodobacter sphaeroides, a purple nonsulfur photosynthetic bacterium, displays exceptional metabolic versatility, enabling growth under both aerobic and anaerobic conditions and utilization of diverse carbon sources. Its flexible metabolism, combined with native pathways for terpenoid and tetrapyrrole biosynthesis, makes it a highly promising microbial chassis for the production of valuable compounds. Advances in metabolic and synthetic biology have allowed the engineering of R. sphaeroides for the efficient synthesis of coenzyme Q10 (CoQ10) and porphyrin derivatives through strategies such as precursor supply enhancement, pathway optimization, modulation of redox and energy balance, manipulation of global regulatory systems, and fermentation control. Beyond CoQ10 and porphyrins, this organism holds the potential to produce hydrogen, carotenoids, and other high-value terpenoids. This review summarizes the metabolic features, native regulatory networks, and engineering approaches in R. sphaeroides, highlighting its versatility and robustness as a platform organism. The insights provided here underscore its potential as a chassis for synthetic biology applications and industrial bioproduction of a wide range of bioactive compounds.
|
|
Scooped by
mhryu@live.com
May 26, 5:18 PM
|
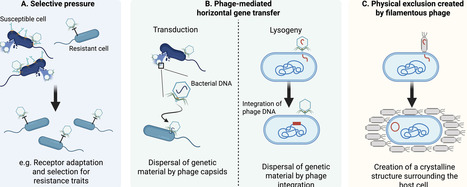
Microbial communities deliver essential functions in ecosystems. In plant environments, the plant microbiome facilitates nutrient uptake, supports plants during abiotic stress, and counteracts disease. As implementation of synthetic microbial communities becomes more of a realistic strategy for mitigating the effects of biotic and abiotic stressors on plant productivity, it is increasingly important to understand how interactions between microbes, which are essential for ecosystem function (hub microbes), are maintained. Recent research highlights the ecological role of bacteriophages, the viruses of bacteria, in host-associated microbial communities. Current evidence demonstrates the influence of the phageome on microbiomes, ranging from effects on an individual (transduction, lysogenic conversion, and evolutionary pressure) to entire populations and communities, such as Kill-the-Winner dynamics. These dynamics appear to affect the overall function of microbial communities and support plant growth. In this review, we lay out recent insights on the role of bacteriophages in plant-associated microbiomes through an eco-evolutionary lens and future directions of research to broaden our understanding of the ecological implications of bacteriophages.
|
|
Scooped by
mhryu@live.com
May 26, 5:09 PM
|
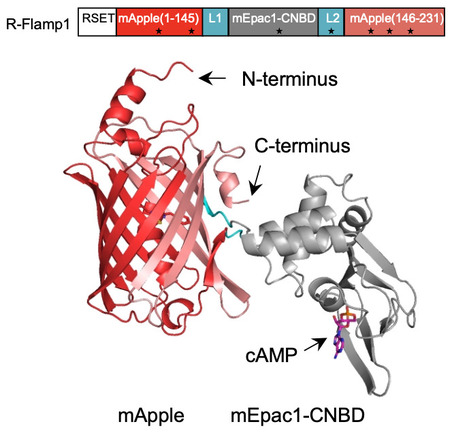
Circularly permuted green fluorescent protein (cpGFP)-based high-performance cAMP sensors have enabled real-time monitoring of cAMP dynamics with high spatiotemporal resolution in living animals. However, their utility is hampered by significant spectral overlap with other green/yellow fluorescent indicators and blue/cyan light-activated optogenetic actuators, limiting their compatibility in multiplexed imaging applications. While existing red cAMP sensors offer great spectral separation, they often suffer from a limited dynamic range ( < 1.5-fold in HEK293T cells), low cellular brightness, aggregation, or significant blue-light-induced photoactivation. Here, we report R-Flamp1, a red cAMP sensor with a large dynamic range ( > 10-fold in HEK293T cells), enhanced cellular brightness, appropriate cAMP affinity (Kd ~1.9 μM), subsecond response kinetics, and minimal photoactivation under blue or cyan light exposure. Using R-Flamp1, we visualized region-specific cAMP dynamics, and when paired with green indicators, revealed differential activation patterns between cAMP and neuromodulators or calcium using two-photon imaging and fiber photometry during various behaviors. These findings provide valuable insights into the role of cAMP signaling in complex behaviors. R-Flamp1 is a high-performance red fluorescent cAMP sensor and the authors monitor region-specific cAMP dynamics in vivo and reveal distinct activation patterns between cAMP, calcium, and neuromodulators via two-photon imaging and fiber photometry in animals.
|
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
Amyloid formation and liquid–liquid phase separation (LLPS) are two important phenomena in cellular biology, linked to both normal physiological functions and various pathologies. Here, we present a computational framework that scores amyloid propensities (amyloid-predict) or LLPS propensities (LLPS-predict) from protein language model embeddings, enabling rapid proteome-wide annotation of peptides and residues. amyloid-predict achieves classification performance that exceeds existing AI and physics-based tools on a hexapeptide benchmark while enabling substantially faster high-throughput screening; notably, amyloid-predict is sensitive to subtle mutational effects and is influenced by sequence patterning and context rather than amino acid composition alone. We apply these protein language model classifiers to all the IDRs in the human proteome and uncover several protein categories with significant enhancement in amyloid and/or LLPS propensity, suggesting insights into the biological roles of these protein categories. For example, signaling receptors, carbohydrate-binding proteins, and Ca2+ binding proteins are enriched in aggregation propensity, while mRNA-binding proteins, ribonucleoprotein complex, and nuclear matrix proteins are enriched in LLPS propensity. Interestingly, we observe patterns of both high amyloid and LLPS propensity in several amyloid-forming and prionic proteins. Together, these results provide side-by-side landscapes of LLPS and amyloid potential across the disordered human proteome while offering a rapid screening tool for basic biology, disease-mechanism studies, and rational design of peptide therapeutics.
|
|
Scooped by
mhryu@live.com
Today, 1:08 AM
|
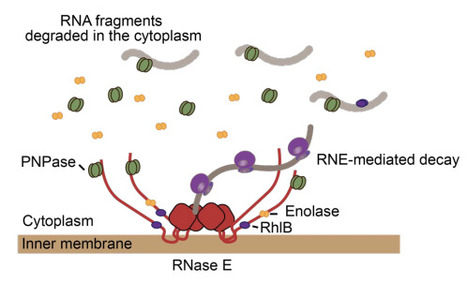
Macromolecular scaffolds are often viewed as fully assembled molecular machines with fixed stoichiometry. Here, we show that the E. coli RNA degradosome follows a more dynamic organizational principle. Using live-cell single-molecule imaging, we quantified the spatial distribution, diffusion, and stoichiometry of RNase E and its accessory factors RhlB, PNPase, and enolase in vivo. The membrane-associated stoichiometry of RhlB and PNPase was broadly consistent with canonical expectation, whereas enolase was underrepresented in the membrane-bound degradosome. In contrast to the near-complete membrane association of RhlB, PNPase partitioned substantially between membrane-associated and cytoplasmic pools, and this partitioning shifted with RNA availability and growth condition. Using lacZ reporters with different translation initiation strengths, we further show that RhlB and PNPase preferentially promote degradation of weakly translated transcripts, whereas strongly translated transcripts are largely insensitive to their loss. Together, these results support a model in which the E. coli RNA degradosome is a membrane-anchored but dynamically assembled complex, with accessory factors contributing differently across physiological states and RNA substrate classes.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Our understanding of protein function and evolution is largely based on the relationship between amino acid sequence and overall fold, now effectively captured by computational models. Yet predicting how mutations—shaped by epistasis—alter protein behavior, especially in dynamic or structurally ambiguous regions, remains difficult. Here we present D2D, which combines a self-supervised protein language model with protein-specific evolutionary information to predict mutational effects using little to no task-specific labeled data. D2D captures long-range epistatic interactions, accurately predicts single and higher-order mutation effects on protein thermostability and binding, without being trained on the task. When fine-tuned, D2D outperforms state-of-the-art methods on latent driver cancer mutations and co-occurring proliferation-enhancing mutations across independent experimental studies. Unlike most existing approaches, D2D avoids biases linked to solvent accessibility or to multiple sequence alignment depth and quality, making it particularly effective for disordered or surface binding regions where structure-based predictors typically falter. Overall, D2D provides a general framework for modeling mutational effects in proteins with limited experimental or structural information.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
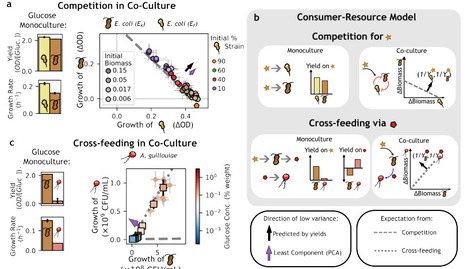
How microbial communities maintain robust and reproducible ecological functions despite their extraordinary taxonomic diversity remains an open question. Here we show that functional organization in microbial communities can be uncovered by repurposing Principal Component Analysis to focus on directions of lowest variance in taxon abundance data, rather than maximal variance. These least-variance components are statistically significant and correspond to ecological constraints on taxon abundances that are consistently fulfilled across samples. Using consumer-resource models, we show that these constraints arise from resource-mediated interactions and express biomass conservation, effectively grouping taxa into producer and consumer guilds. We validate this interpretation in simulated communities and experimental systems under competition and cross-feeding. Finally, we show that low-variance structure is ubiquitous in natural microbial communities and reveals a sparse network of taxa with disproportionate influence on community structure. Together, our results establish low-variance components as indicators of ecological constraints linking taxonomic diversity to functional organization.
|
|
Scooped by
mhryu@live.com
May 26, 10:35 PM
|
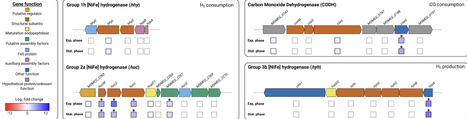
Certain hydrogenases are capable of converting the trace amounts of molecular hydrogen constantly present in Earth’s atmosphere to supply cells with a little energy during periods of starvation. Recently, Kropp and colleagues investigated the regulation of one of these “high-affinity” hydrogenases in response to different growth conditions for the ubiquitous soil bacterium Mycobacterium smegmatis (A. Kropp, J. D. Archer, M. Jespersen, T. D. Watts, et al., mSystems 11:e01678-25, 2026, https://doi.org/10.1128/msystems.01678-25). A mutation in the gylR gene, whose product acts as a positive transcriptional regulator of the genes involved in glycerol catabolism, causes the corresponding mutant strain to grow very slowly on glycerol, while the activity of the high-affinity hydrogenase reached levels more than 50 times higher than those of the wild-type strain of M. smegmatis. The results of this study suggest that the synthesis of the hydrogenase is subject to a regulatory mechanism similar to carbon catabolite repression, which is entirely consistent with the cellular function of this enzyme.
|
|
Scooped by
mhryu@live.com
May 26, 7:22 PM
|
Faster drug discovery, optimized drug design, even programmable therapeutics: AI is impacting R&D. Data and development bottlenecks remain. Some of 2026’s largest IPOs, deals and venture rounds are AI-powered. Generate Biomedicines pulled off a $400 million IPO in February 2026 for its protein generation platform. Eli Lilly in March paid $115 million up front for a drug discovery partnership with Insilico Medicine, itself hot off the back of a $293 million Hong Kong listing. In the first three months of 2026, AI and machine learning (ML)-based drug discovery firms raised $1.8 billion in venture funding, about a quarter of the sector total. That included an $80 million seed round for New York and Boston-based Proxima, whose plans include reprogramming proteins and “decoding the structure of life.”
|
|
Scooped by
mhryu@live.com
May 26, 6:55 PM
|
Hydroponic horticulture will play a key role in future food production as the growing global population becomes increasingly urbanised. Tomato (Solanum lycopersicum) is a widely grown and consumed crop that is already cultivated hydroponically in glasshouses in areas of the world with cooler climates, such as Northern Europe. Hydroponic growing systems enable high yields but can enhance disease susceptibility which increases the risk of devastating yield losses. Manipulation of the hydroponic microbiome has been proposed as a strategy to protect plants against disease. However, this hypothesis remains largely untested. We examined whether introducing synthetic communities of plant-beneficial microbes (SynComs) could offer a sustainable disease protection solution for hydroponic tomato production. We identified individual microbes and in turn two SynComs that induce systemic disease resistance during the vulnerable early stages of development. The two SynComs were evaluated further in a commercial-scale greenhouse trial. Although both SynComs reduced early growth, they had no adverse effects on yield or fruit quality. Strikingly, while only one SynCom strain consistently persisted in the hydroponic stone wool substrate throughout the six-month trial, the introduction of disease-suppressive SynComs at sowing had significant and similar impacts on bacterial community structure six months later. Our findings demonstrate that microbial SynComs can reduce disease susceptibility of hydroponically grown tomato without compromising yield, offering a viable and sustainable approach for crop protection in controlled environment agriculture.
|
|
Scooped by
mhryu@live.com
May 26, 5:55 PM
|
Plasmids drive evolution by transferring traits across microbial hosts. Transmission depends on both host–plasmid (infection) and plasmid–plasmid (compatibility) interactions, yet how the structure of these networks shapes transmission remains poorly understood. We hypothesized that these two ecological networks interact in non-additive ways to influence community outcomes. To test this, we developed a stochastic agent-based model that embeds both network structures and simulates coupled host–plasmid dynamics. We systematically varied the structure of each network, both individually and in combination, to isolate the effect of structure on host-plasmid dynamics. A modular (interactions organized into clusters) and hub (interactions concentrated on the highly connected) plasmid-plasmid compatibility network promoted transient host coexistence, while a modular host-plasmid infection network promoted plasmid diversity and stable host coexistence. Importantly, structured networks interacted non-additively, and their impact was most apparent when plasmid carriage imposed a moderate fitness cost on hosts. For example, combining a modular infection network with a hub compatibility network reversed the expected plasmid prevalence patterns, demonstrating that the structure of one network can counteract the effects of the other. We further re-parameterized our model to recapitulate empirical host-plasmid community dynamics, showing that infection network structure can strongly shape plasmid prevalence even in the presence of substantial biological heterogeneity. Our results highlight the necessity of jointly considering host–plasmid infection and plasmid–plasmid compatibility networks to understand host–plasmid community dynamics and their eco-evolutionary potential. More broadly, this work provides an initial mechanistic framework for generating testable hypotheses and underscores that systems involving multiple hosts and infectious agents require explicit consideration of how different ecological networks interact to shape community dynamics.
|
|
Scooped by
mhryu@live.com
May 26, 5:46 PM
|
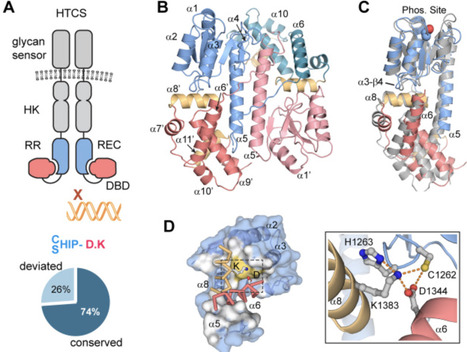
Human gut microbes, such as Bacteroides, rely on specialized gene clusters known as polysaccharide utilization loci (PULs) to metabolize diverse dietary and host-derived glycans. A major class of transcription regulators of these PULs is the hybrid two-component system (HTCS) containing a histidine sensor kinase and a response regulator (RR) within a single transmembrane polypeptide chain. Characterizing HTCS-mediated PUL regulation is often challenging because the specific glycan signals required to activate most HTCSs remain unknown. Here, we characterized structural details of a highly conserved HTCS activation mechanism and developed a universal activation strategy by mutating the interdomain latch motif that inhibits the DNA-binding activities. Using the RR portion of BT4124 from Bacteroides thetaiotaomicron as a model system, crystallographic analyses reveal a “closed” inactive conformation anchored by a hydrogen-bond network formed by the conserved latch residues between the receiver and DNA-binding domains. Molecular dynamic simulation with the deep-learning BioEmu shows that the “AD” mutation of the latch residues destabilizes the inhibitory interface, shifting the conformation equilibrium predominantly to an active, “open” conformation. This constitutively active variant, BT4124RAD, allows us to map specific DNA-binding sites within the potential regulated promoters in vitro and characterize transcription regulation in cells. Induced expression of BT4124RAD not only down-regulates local homogalacturonan (HG) utilization genes but also cross-represses multiple PULs associated with other HG-related pectic glycans. These findings highlight a complex cross-regulatory network governing pectin degradation and establish the targeted latch mutation as a potential broadly applicable tool for deciphering the regulatory networks of HTCSs in Bacteroides.
|
|
Scooped by
mhryu@live.com
May 26, 5:28 PM
|
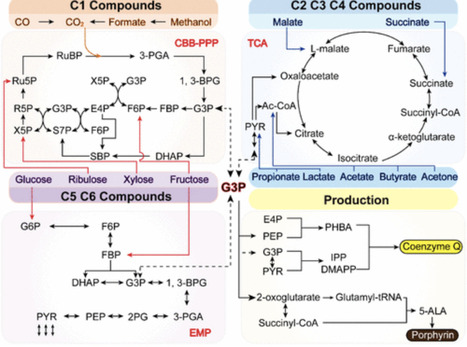
Methanol has emerged as a sustainable C1 feedstock owing to its compatibility with existing infrastructure and the potential for renewable production from CO₂ and green hydrogen. Methylotrophic yeasts, including Komagataella phaffii (Pichia pastoris) and Ogataea polymorpha, can natively assimilate methanol and therefore represent attractive hosts for biomanufacturing. However, industrial application of methanol-based processes remains constrained by cytotoxicity, redox imbalance, and limited productivity compared to sugar-based fermentations. To address these challenges, extensive metabolic engineering strategies have been implemented to enhance methanol assimilation and redirect carbon flux toward value-added products. Over the past decade, remarkable progress has been achieved through the development of synthetic methylotrophy in non-methylotrophic yeasts, the expansion of product portfolios to glycans, fatty acid derivatives, polyketides, terpenoids, organic acids, and polyols, and the integration of multi-omics tools for systems-level design. This review summarizes recent advances in methanol assimilation enhancement, synthetic pathway construction, and fermentation engineering, highlighting strategies such as metabolic engineering and dynamic bioprocess control. In addition, current challenges and future perspectives are discussed with an emphasis on overcoming toxicity, improving efficiency, and establishing advanced methylotrophic yeasts as robust cell factories for sustainable C1-based biomanufacturing.
|
|
Scooped by
mhryu@live.com
May 26, 5:21 PM
|
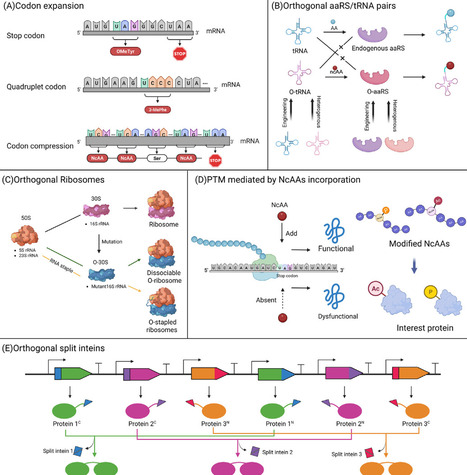
Precise regulation of protein abundance is essential for cellular function and physiology. Conventional approaches are often limited by insufficient resolution or unintended crosstalk. In contrast, orthogonal control technologies enable programmable and precise modulation of protein abundance while remaining insulated from native networks. In this review, we summarize the development and application of regulation technologies with different orthogonality across multiple levels. Orthogonal transcriptional control primarily involves the design and engineering of orthogonal RNA polymerases and transcription factors; orthogonal translational regulation focuses on advances in genetic codon expansion and post-translational modifications; targeted protein degradation and compartmentalized regulation are also discussed. Finally, we highlight the integration across the different levels described above. This review might bring disruptive insights and conceptual breakthroughs to precision medicine and sustainable biomanufacturing.
|
|
Scooped by
mhryu@live.com
May 26, 5:16 PM
|
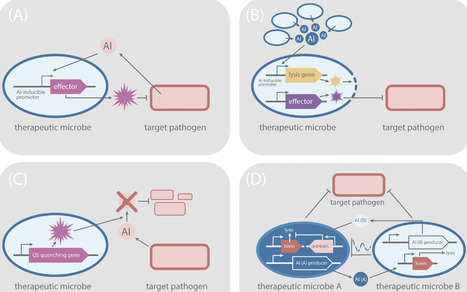
Quorum sensing (QS) is a cell–cell communication mechanism that enables bacteria to coordinate gene expression in response to population density and community composition. In many pathogens, QS plays a central role in host colonization and virulence, making it an attractive target for antimicrobial intervention. Synthetic biology offers powerful tools to exploit this vulnerability by either disrupting QS signaling or engineering microorganisms with QS-based circuits to detect and eliminate pathogens. In this review, we examine how QS and QS interference can be harnessed for QS circuit engineering and translated into applications such as therapeutic microorganisms. We also highlight the transition of QS research from fundamental microbiology to translational biotechnology, underscoring its potential to drive innovative strategies against microbial virulence and antimicrobial resistance.
|





















persistence, colonization