 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:51 AM
|
Metagenomes offer the potential to characterize E. coli strain-level diversity within the human gut microbiome, informing our understanding of colonization diversity and the genetic features distinguishing infection from carriage. Among numerous reference-based tools for short-read metagenomic strain-level profiling, the best approach remains unclear. Here, we benchmarked six published tools—PanTax, PathoScope, StrainGE, Strainify, StrainR2 and StrainScan—for their ability to detect co-existing strains of E. coli and estimate their relative abundance across real and simulated metagenomes of increasing complexity with varying reference database composition. In the ZymoBIOMICS® D6331 dataset, only PanTax achieved zero error when predicting the equal abundance of five E. coli strains. In a differentially abundant four-strain mock community dataset (SRR13355226), StrainScan had the lowest mean absolute proportional error (0.89), driven by reduced sensitivity (0.5), followed by PathoScope (4.08). Across simulated metagenomes reflecting the healthy adult gut microbiome, all tools demonstrated high sensitivity (≥0.833), but specificity, precision and F1 score were selectively improved in some tools through detection thresholds to remove low abundance false positives. Outright, StrainGE achieved the highest F1 score (0.978). Predicted relative abundances of the E. coli K12-MG1655 (phylogroup A) and O157:H7 Sakai (phylogroup E) strains spiked into simulated metagenomes across varying abundance ratios were generally accurate, with PanTax and StrainR2 showing the lowest mean absolute proportional error (0.06). When truly present strains were removed from the reference database, out-of-phylogroup assignments were observed for some tools. Collectively, our results demonstrate that published metagenomic strain-level profiling tools vary in their ability to profile E. coli strains, indicating that method selection should be guided by intended application. These findings will facilitate characterisation of E. coli strain-level diversity within short-read gut metagenomes with greater accuracy than previously possible.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Whether and how root exudates of Amorpha fruticosa Linn. mediate the positive effects of microbial inoculation (Bacillus thuringiensis NL-11 and Gongronella butleri NL-15) on soil aggregation remains unclear. We conducted two glasshouse experiments and a controlled laboratory incubation experiment to address this knowledge gap. Glasshouse experiments showed that microbial inoculation increased root exudation, changed soil microbial community structure, and enhanced soil aggregation and mean weight diameter (MWD). Microbial inoculation increased the number of nodes and links in bacterial–fungal exudate association networks across soil aggregate sizes. Network properties, including the number of nodes, total links, and microbial–exudate (ME) links, were positively correlated with soil MWD and mineral-associated organic carbon (MAOC). Partial least squares path modeling revealed that inoculation indirectly increases MWD by enhancing MAOC, which subsequently strengthens ME links. The laboratory incubation experiment further showed that a single addition of inoculation-induced exudates did not promote macroaggregate formation but significantly increased MAOC content relative to the control. Inoculation-induced root exudates increased the number of nodes and links in bacterial–fungal association networks across all aggregate sizes, and these network attributes were positively correlated with MAOC. We demonstrate that microbial inoculation enhances soil aggregation and carbon stabilization through root exudate-mediated microbial association networks.
|
|
Scooped by
mhryu@live.com
Today, 12:33 AM
|
Salinity disrupts ionic balance, osmotic regulation, redox homeostasis, and hormone signaling, thereby constraining plant growth and metabolism. Plants employ adaptive responses to salinity stress, including ion transport regulation, compatible solute accumulation, antioxidant defenses, and signaling reprogramming; however, these mechanisms are often insufficient under high salinity. Beneficial microbes, including plant growth-promoting rhizobacteria, endophytes, fungi, and halotolerant taxa, are increasingly associated with improved plant performance under saline conditions. These associations are consistently linked with changes in ion homeostasis, osmotic adjustments, redox balance, hormone signaling, and root architecture. Importantly, most available evidence derives from transcriptomic, biochemical, and physiological observations, which do not establish direct mechanistic regulation. This review critically evaluates microbial contributions to plant salinity responses by explicitly distinguishing between experimentally validated mechanisms, correlative associations, and hypothesis-driven models. We integrate insights from molecular genetics, biochemistry, and multi-omics approaches while highlighting their limitations in establishing causality. Particular emphasis is placed on experimental strategies required to establish causal mechanisms, including isotope tracing, genetic perturbation, and synthetic community approaches.
|
|
Scooped by
mhryu@live.com
Today, 12:19 AM
|
Precise placement of the cell division machinery is essential for cell division in most organisms, yet the mechanisms responsible for this process vary substantially across the domains of life. In Eukaryotes, cell division is primarily driven by ESCRT-based systems, while many Bacteria rely on the Min system to ensure accurate positioning of the division septum. By contrast, the mechanisms by which Archaea spatially regulate divisome assembly remain largely unknown. Here, we identify a three-protein system, which we term divisome positioning proteins A, B and C (DipA, DipB and DipC) that is essential for correct divisome positioning in Haloferax volcanii. Deletion of any dip gene results in mislocalization of cell division proteins and the formation of abundant minicells, similar to Min defects in bacteria. Furthermore, we show that all three Dip proteins undergo pole-to-pole oscillation, and that DipB and DipC assemble into ring-like structures at midcell. Biochemical analyses demonstrate that DipA is a membrane binding GTPase whose association with the membrane is disrupted by DipB. Notably, Dip homologues are widely conserved across diverse archaeal lineages that employ FtsZ-based division, indicating that the Dip system is a broadly distributed key regulator of FtsZ-based cell division in Archaea. Despite its striking functional similarities to the bacterial Min system, the Dip system is entirely unrelated at the sequence level, representing a compelling example of convergent evolution of oscillatory spatial regulators in distinct domains of life.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
Cable bacteria are filamentous sulphide-oxidizing bacteria that transport electrons through conductive periplasmic fibres over centimetre-scale distances in redox-stratified sediments to respire oxygen. Here, we show that the freshwater cable bacterium Electronema aureum GS employs a versatile extracellular electron transfer (EET) system to respire insoluble electron acceptors under anoxic conditions, achieving growth rates comparable to aerobic respiration. Electrochemical and molecular analyses reveal that E. aureum GS performs both direct and riboflavin-mediated EET to electrodes, including at +600 mV vs. Ag/AgCl, an unusually high redox potential typically not accessed by electroactive bacteria. Two distinct cell-surface redox components were identified, which are metal-dependent, pH-sensitive, and heat-labile, consistent with outer-membrane-localized cytochromes. The redox shuttle riboflavin accumulated in bioelectrochemical systems with E. aureum GS, supporting mediated EET. These findings reveal a respiratory flexibility in E. aureum GS and highlight the role of EET in energy conservation in dynamic redox environments. In this study, the authors show that cable bacteria perform extracellular electron transfer via direct and flavin-mediated pathways, which enables growth under anoxic conditions at rates comparable to aerobic respiration, revealing their metabolic versatility.
|
|
Scooped by
mhryu@live.com
May 19, 11:58 PM
|
Plant intelligence is defined as the capacity of plants to integrate environmental signals through distributed biochemical, electrical, and epigenetic networks, resulting in adaptive physiological or developmental responses without requiring neural cognition. This definition distinguishes plant adaptive plasticity from animal cognition while maintaining a rigorous biological framework. This phenomenon has captivated biologists for decades, yet its underlying mechanisms remain elusive amid intricate biological interactions and vast, complex datasets. Recent breakthroughs in synthetic biology and artificial intelligence (AI) offer unprecedented opportunities to decode these adaptive strategies in silico. This review explores hybrid AI systems that integrates predictive AI, agent-based modelling, including large language models (LLMs) and retrieval-augmented generation (RAG), and bio-inspired algorithms, charting innovative pathways for simulating and investigating plant adaptive behaviours via synthetic biology. We highlight how these hybrid approaches bridge empirical science and theoretical insights, empowering the design of resilient synthetic biological systems that emulate plants' extraordinary adaptability to diverse challenges.
|
|
Scooped by
mhryu@live.com
May 19, 11:46 PM
|
Synonymous mutations, long regarded as evolutionarily neutral, are increasingly recognized as important determinants of viral evolution and adaptation. In this review, we propose that viral codon usage reflects a multi-objective optimization process shaped by competing evolutionary constraints, positioning synonymous variation as a key layer of translational control with direct implications for viral evolution and rational vaccine design. We first summarize the quantitative metrics used to characterize viral codon usage bias and then examine the evolutionary forces that shape viral codon usage profiles. In particular, we discuss growing evidence that viral codon usage frequently diverges from host preferences, reflecting mutational bias, trade-offs among translational efficiency, immune pressure, RNA structure, co-translational protein folding, and transmission between hosts, rather than simple optimization. We then focus on strategies that deliberately manipulate codon usage for vaccine development. Finally, we discuss the current challenges and how emerging artificial intelligence-based approaches may enable more predictive and integrative codon engineering strategies.
|
|
Scooped by
mhryu@live.com
May 19, 11:26 PM
|
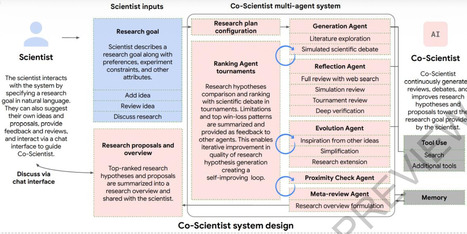
Scientific discovery is driven by scientists generating novel hypotheses for complex problems that undergo rigorous experimental validation. To augment this process, we introduce Co-Scientist, a multi-agent AI system built on Gemini for structured scientific thinking and hypothesis generation. Co-Scientist aims to help scientists discover new original knowledge. Conditioned on their research objectives and prior scientific evidence, it formulates demonstrably novel research hypotheses for experimental verification. The system’s design involves agents continuously generating, critiquing and refining hypotheses accelerated by scaling test-time compute. Key contributions include: (1) a multi-agent architecture with an asynchronous task execution framework for flexible compute scaling; (2) a tournament evolution process for self-improving hypotheses generation. Automated evaluations show continued benefits of test-time compute scaling, improving hypothesis quality over time. While general purpose, we focus the validation in three biomedical applications: drug repurposing, novel target discovery, and explaining mechanisms of anti-microbial resistance. Specifically, Co-Scientist helped identify new drug repurposing candidates and synergistic combination therapies for acute myeloid leukemia, which were validated through in vitro experiments. These real-world validations demonstrate the potential of Co-Scientist to accelerate scientific discovery and usher in an era of AI empowered scientists.
|
|
Scooped by
mhryu@live.com
May 19, 5:51 PM
|
Organic compounds with a negative nominal oxidation state of carbon (NOSC) are thermodynamically recalcitrant in anaerobic ecosystems, but few studies have measured the influence of NOSC on carbon degradation rates, gaseous product yields, or microbiome composition. We amended anaerobic rice paddy sediment microcosms with water-soluble monomeric organic carbon compounds varying in NOSC. Consistent with thermodynamic and stoichiometric predictions, negative NOSC compounds are catabolized more slowly but produce more methane per mole of carbon. Negative NOSC microbiomes have higher alpha diversity, more syntrophs and methanogens, and fewer fermentative bacteria. Strikingly, fermentative bacterial taxa display genomically encoded NOSC catabolic preferences both in the lab and field. Negative NOSC-preferring fermenters have longer predicted doubling times, consistent with the thermodynamic recalcitrance of their preferred substrates. We propose that microbial NOSC catabolic preferences may reflect the thermodynamic niche of microorganisms and we anticipate that extending research on microbial catabolic preferences to a greater variety of organic carbon substrates and diverse microbiomes will improve our understanding of microbial carbon cycling and trait evolution. Microbial methanogenesis is responsible for ~70% of global methane emissions. Here, the authors show that negative nominal oxidation state of carbon (NOSC) compounds are catabolized more slowly but produce more methane per mole of carbon and that NOSC impacts microbial community composition in microcosms.
|
|
Scooped by
mhryu@live.com
May 19, 5:36 PM
|
The rapid explosion of large-scale, high-throughput biological data has created an urgent demand for efficient analysis pipelines. Traditional bioinformatics approaches, while powerful, often require specialized computational expertise, placing them out of reach for bench biologists. Large Language Models (LLMs) offer new possibilities for automating complex reasoning and tool integration, yet existing LLM-based solutions have not sufficiently lowered this barrier, and expert-level analysis remains inaccessible to most nonexperts. Here, we present BioGAIP, an LLM-powered agent that integrates expert-level reasoning within an end-to-end platform for bioinformatics tasks. By coupling optimized autonomous agents with full graphical interfaces, BioGAIP transforms complex analytical workflows into an automated, user-friendly, and low-intervention process with natural language input. Key features of BioGAIP include dynamic information retrieval, automatic environment configuration, and self-directed design of analysis pipelines, making large-scale multi-omics analysis highly accessible. Built on agent-based client-server architecture, BioGAIP ensures secure resource management and supports heavy computational demands. Extensive evaluations on diverse published datasets demonstrate that BioGAIP reliably recapitulates established biological insights and shows strong potential for novel discovery. By democratizing complex bioinformatics workflows, BioGAIP accelerates accessible data-driven discovery for both experts and nonexperts.
|
|
Scooped by
mhryu@live.com
May 19, 2:53 PM
|
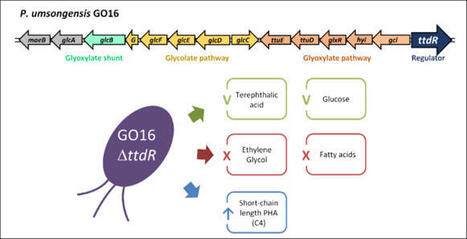
In Pseudomonas umsongensis GO16, a LysR-type transcriptional regulator (LTTRs) ttdR is located 5′ of the gene for glyoxylate carboligase (gcl), involved in ethylene glycol (EG) metabolism. GO16 wildtype (WT) can utilize EG as a sole carbon and energy source but the ttdR knockout strain (ΔttdR) cannot grow on EG, demonstrating its role as an activator in EG metabolism. When ΔttdR was grown with terephthalic acid (TA) or glucose, the growth was comparable to the WT. GO16 originally produces both C4 short-chain-length (scl) PHA and C6-C12 medium-chain-length (mcl) PHA from various carbon sources. Interestingly, ΔttdR produced scl-PHA monomer 5.4-fold and 1.4-fold increased than the WT when TA and glucose were used respectively. Furthermore, ΔttdR exhibited a very long lag phase when grown with fatty acids, namely butyrate or octanoate. This indicates a more complex role of TtdR than simply activating EG metabolism in GO16. The analysis of the GO16 ΔttdR proteome revealed that EG catabolic genes were expressed, but their abundance was 2.8- to 10.5-fold lower (log2) compared to the WT. In addition, higher abundance of enzymes that could be contributing to higher scl-PHA content was also observed. GO16 ΔttdR was further subjected to adaptive laboratory evolution (ALE) with butyrate and octanoate. The evolved strains regained the ability to grow with these fatty acids as the WT, but the mutations accumulated did not confer growth with EG or acetate, suggesting that a mechanism different to glyoxylate shunt has evolved to allow growth with fatty acids.
|
|
Scooped by
mhryu@live.com
May 19, 12:41 PM
|
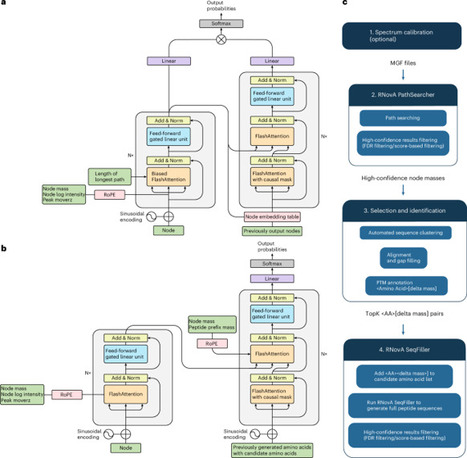
De novo peptide sequencing directly infers sequences from mass spectrometry data without relying on protein databases. Although recent deep learning models can also identify posttranslational modifications (PTMs), they require labeled training data for this task. Here we introduce rotary positional embedding-enhanced de novo sequencing algorithm (RNovA), a transformer-based de novo sequencing algorithm enhanced with relative positional embeddings and a reinforcement-learning-style sequential decision framework. RNovA enables open PTM discovery in a zero-shot setting—without retraining or a predefined list of candidate residues—while maintaining state-of-the-art performance on standard benchmarks. Demonstrating this capability, we successfully identified peptides modified by kynurenine—an uncommon and biologically relevant PTM—in clinical samples from patients with RA and validated this discovery with synthetically synthesized reference peptides. Furthermore, we demonstrated open de novo PTM discovery by analyzing the bacterial strain A1232E, which lacks a reference proteome, and detected an unannotated glutamic acid modification. RNovA enables exploration of previously inaccessible regions of the proteome, including peptides with unexpected or unannotated modifications. RNovA is an open-search de novo peptide sequencing model.
|
|
Scooped by
mhryu@live.com
May 19, 11:49 AM
|
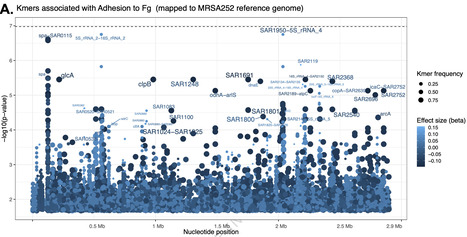
Staphylococcus aureus is a major cause of bloodstream infections, but how variation between strains in their ability to adhere to host proteins influences disease severity remains unclear. Here we show that adhesion is a highly variable and biologically important trait in 236 phylogenetically diverse, representative bacteremia isolates profiled for binding to fibrinogen and fibronectin and analysed together with bacterial whole-genome sequences and matched patients’ clinical data. Stronger fibrinogen-binding, particularly in strains lacking α-toxin, correlates with heightened systemic inflammation (r = 0.401, P = 0.0001) but lower mortality (16.6% vs. 38.7%, P = 0.018), linking bacterial adhesion to distinct clinical outcomes. Genome-wide association analyses identify top-associated variants in genes encoding known adhesins (clfA, fnbA, fnbB, ebh), other surface factors (spa, sdrH), mobile genetic elements, and novel loci (csbB, glcA), although not statistically significant. Functional analyses reveal that protein A limits ClfA-dependent fibrinogen binding through steric hindrance, CsbB interferes with ClfA exposure on bacterial surface, and GlcA enhances fibronectin binding via metabolic regulation. These findings define adhesion as a polygenic, evolutionarily variable trait and suggest that highly-adhesive, α-toxin-defective isolates promote inflammatory but self-limiting infections, whereas weakly adhesive, high-toxicity strains favour immune evasion and severe disease. To uncover how Staphylococcus aureus strain variation influences adhesion and thus disease outcomes, the authors profile 236 bacteremia isolates, showing that high-adhesion, low-toxicity isolates may elicit inflammatory yet self-limiting infections.
|
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
Soil salinization, a growing global issue threatening sustainable agriculture, can be mitigated by Plant Growth-Promoting Bacteria (PGPB) as a green strategy. PGPB primarily consist of plant growth-promoting rhizobacteria (PGPR) and plant growth-promoting endophytes (PGPE). Numerous studies have demonstrated that both types of bacteria can enhance plant performance under salt stress through various mechanisms that help maintain ion homoeostasis, improve osmotic adjustment, and enhance antioxidant defence. Although PGPR and PGPE share highly conserved core mechanisms, their distinct colonization niches lead to functional divergence in efficacy, stability, and ecological roles. This review systematically summarizes these conserved mechanisms for enhancing plant salt tolerance. Furthermore, it elaborates on their functional differences across various ecological niches and discusses the major challenges and future directions for the field application of these PGPB. Ultimately, this review aims to provide a theoretical foundation for the scientific deployment of PGPB in saline agroecosystems.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
Rhizosphere metabolites play a pivotal role in plant–soil–microbe interactions, yet the absence of standardized sampling method presents challenges for data comparability and a unified understanding of rhizochemistry dynamics. In this study, the rhizosphere metabolites of spring wheat (Triticum aestivum L.) were systematically evaluated using three commonly used sampling methods – rhizosphere soil extraction, hydroponic collection, and in situ sampling. We assessed the differences in metabolite abundance, diversity, composition, and metabolic pathways among the three methods. The results revealed that rhizosphere soil extraction method is simple and repeatable, but it tends to underestimate the rare metabolic features compared to the other two methods. The hydroponic collection method offered a more comprehensive rhizochemical profile, however, it may not reflect the actual soil environment in which plants grow. The in situ method mostly captured the rhizochemical dynamics under natural conditions but was technically complex and less reproducible. Pathway enrichment of differential metabolic features showed a distinct pattern, with hydroponic emphasising plant-centric primary metabolism, and rhizosphere soil extraction and in situ methods reflecting pathways more associated with environmental interaction and ecological adaptation. Overall, our results highlight that the sampling methods significantly influences the abundance and composition of the detected rhizochemical profile, thus the rhizosphere metabolites sampling methods should be considered according to the experimental objectives of future research.
|
|
Scooped by
mhryu@live.com
Today, 12:30 AM
|
TALEs (transcription activator-like effectors) are an excellent example of how studying pathogen–host interactions can lead to significant biotechnology inventions. TALEs are bacterial effectors that are translocated into plant cells via a bacterial type III secretion system. Once inside the host cell, they are imported into the nucleus to bind specific promoters and induce expression of target genes, thereby supporting the bacterial infection. TALEs are found throughout many, but not all, Xanthomonas pathovars, which can be severe pathogens of different crops. The key feature of TALEs is their modular DNA-binding domain, which allows a simple evolutionary adaptation to novel DNA sequences as well as simple cloning of designer TALEs with desired DNA-binding specificity. Accordingly, TALE-nucleases started the genome-editing revolution, and TALE base editors are the latest tools to efficiently edit chloroplast and mitochondrial genomes. We review recent advances in Xanthomonas genomics, synthesize current knowledge about naturally occurring TALEs, and highlight current roles of TALEs in genome editing and synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 12:16 AM
|
Arbitrium is a communication system that helps bacteriophages to decide between lysis and lysogeny through secreted peptides. In this system, the arbitrium communication peptide (AimP) binds its cognate arbitrium receptor (AimR) to repress aimX (a negative regulator of lysogeny) expression, promoting lysogeny. It has been assumed that each AimR responds exclusively to its own AimP. Here, we challenge this view by demonstrating cross-communication between arbitrium systems. Using prototypical arbitrium phages, we show that AimP peptides can bind and repress non-cognate AimR receptors, promoting lysogeny and reducing prophage induction. Structural and biochemical analyses reveal conserved receptor features that permit cross-recognition of non-cognate peptides while preserving recognition of cognate partners. In mixed lysogenic cultures, these interactions alter induction outcomes, underscoring their ecological significance. Extending to infection contexts, we demonstrate that crosstalk favors lysogeny of incoming phages in cells harboring compatible systems. These findings establish that phages engage in cross-species communication via peptide signaling, reshaping microbial communities in unexpected ways.
|
|
Scooped by
mhryu@live.com
Today, 12:03 AM
|
Bacteria dominate the biosphere and assemble into highly diverse communities, yet the mechanisms by which these communities migrate remain poorly understood. Here, we used a meso-tube chemotaxis assay to track taxonomic, functional and genomic shifts within sewage-derived microbial communities that migrate in self-organized bands over metre scales. Chemotactic bands accelerated during migration and incorporated non-motile bacterial hitchhikers as well as up to 10⁶ viruses/mL. Approximately 500 species co-migrated, with relative abundances fluctuating by orders of magnitude over time. The final communities exhibited enrichment of functional genes linked to motility and chemotaxis, consistent with adaptation to migration. Despite this functional convergence, replicate communities differed in taxonomic composition, reflecting environmental filtering that selects for functionally equivalent species. By revealing how chemotaxis governs large-scale microbial migration, this work provides a framework for understanding microbial spread in natural ecosystems and host-associated environments. Migration of microbial communities is poorly understood. Here, the authors use a meso-tube assay to show that hundreds of microbial species co-migrate over metre scales via chemotaxis, which restructures communities, enriches motility traits and facilitates dispersal of viruses and non-motile ‘hitchhikers’.
|
|
Scooped by
mhryu@live.com
May 19, 11:56 PM
|
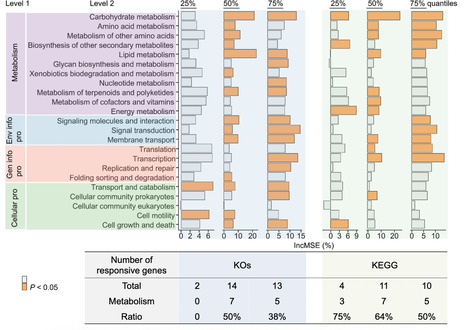
Microbial carbohydrate-active enzymes (CAZymes) underpin carbon cycling across Earth’s ecosystems; however, how contrasting environments shape CAZyme diversity and structural conservation remains poorly understood. Here, we applied shotgun metagenomics to compare the carbohydrate-degradation potential of two functionally prolific but physicochemically opposed ecosystems: the alkaline-saline soda lakes of the East African Rift Valley and the anaerobic ruminant gut. From 34 metagenomes (12 soda lake and 22 rumen), we recovered 371 quality-filtered metagenome-assembled genomes, of which 84% of soda lake and 52% of rumen MAGs represented novel species. Rumen communities, dominated by Bacteroidota, Fibrobacterota, and Bacillota, exhibited significantly higher taxonomic diversity and were enriched in carbohydrate catabolism and fermentation pathways. Soda lake communities, dominated by Pseudomonadota, displayed greater evolutionary divergence (lower RED scores) and were enriched in pH homeostasis, oxidative and osmotic stress, sulfur cycling, and carbon fixation pathways. To assess whether structural conservation persists despite extreme sequence divergence, we predicted three-dimensional structures for 12 representative enzymes from six glycoside hydrolase families (GH1, GH3, GH5_11, GH9, GH10, and GH28) using AlphaFold 3. All 12 structures adopted canonical GH family folds with high confidence (pTM 0.75–0.97). These results demonstrate that environmental selection drives distinct taxonomic and functional strategies for carbon processing while preserving three-dimensional CAZyme architecture, positioning soda lake and rumen metagenomes as complementary reservoirs for bioprospecting industrially relevant enzymes.
|
|
Scooped by
mhryu@live.com
May 19, 11:34 PM
|
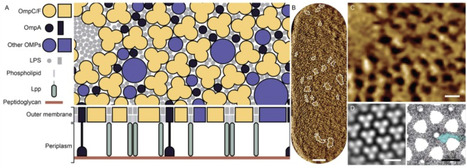
Bacteria can adopt a variety of strategies to shield themselves from environmental insult. Among these, the outer membrane (OM) and S-layers are both surface-exposed layers with overlapping functions. Despite their existence and composition being described decades ago, recent advances have led to a revised view of their biological functions, structural organisation and spatiotemporal dynamics. Here, we review recent advances in our understanding of these envelope layers. We focus on new insights into the biological significance, molecular composition, surface architecture and spatiotemporal patterns underlying their biogenesis. By comparing these systems, we show that the OM and the S-layer share notable features, including aspects of surface symmetry and the spatiotemporal coordination of their assembly. We discuss outstanding questions related to each of these envelope layers and the potential interplay between them. Understanding this interplay promises to reveal fundamental principles of bacterial cell envelope assembly and organisation, opening new avenues for antimicrobial targeting.
|
|
Scooped by
mhryu@live.com
May 19, 11:24 PM
|
The cycle of scientific discovery is frequently bottlenecked by the slow, manual creation of software to support computational experiments. To address this, we present Empirical Research Assistance (ERA), an AI system that creates expert-level scientific software whose goal is to maximize a quality metric. The system uses a Large Language Model (LLM) and Tree Search (TS) to systematically improve the quality metric and intelligently navigate the large space of possible solutions. ERA achieves expert-level results when it explores and integrates complex research ideas from external sources. The effectiveness of tree search is demonstrated across a diverse range of tasks. In bioinformatics, ERA discovered 40 novel methods for single-cell data analysis that outperformed the top human-developed methods on a public leaderboard. In epidemiology, ERA generated 14 models that outperformed the CDC ensemble and all other individual models for forecasting COVID-19 hospitalizations. ERA also produced expert-level software for geospatial analysis, neural activity prediction in zebrafish, and numerical solution of integrals, and a novel rule-based construction for time series forecasting. By devising and implementing novel solutions to diverse tasks, ERA represents a significant step towards accelerating scientific progress.
|
|
Scooped by
mhryu@live.com
May 19, 5:43 PM
|
Microbes, as the planet’s most abundant and diverse organisms, drive soil functions globally and are vulnerable to environmental stressors triggered by global change. Yet, knowledge regarding the impacts of multiple environmental stressors on their functional profiles as well as the consequences for soil functionality largely remains unknown. Here, we analyze two global-scale datasets including information on soil metagenomics and multiple environmental stressors. We find that across terrestrial ecosystems worldwide, up to 60% of all functional genes significantly shift when soil microbes experience the high-level of concurrent stressors. In this regard, the relative abundances of genes involved in microbial growth are negatively linked to the increasing number of stressors. Conversely, those genes linked to stress resistance and energy production exhibit positive responses. Taken together, our findings highlight a significant restructuring of global soil functional microbiomes in response to multiple environmental stressors. Consequently, such restructuring drives community-level shifts in matter and energy reallocations, thereby impacting the maintenance of soil functionality under the projected global change. Soil microbes drive ecosystem functions but are vulnerable to environmental stressors triggered by global change. This study reveals that multiple environmental stressors drive community-level restructuring of soil functional microbiomes globally.
|
|
Scooped by
mhryu@live.com
May 19, 2:54 PM
|
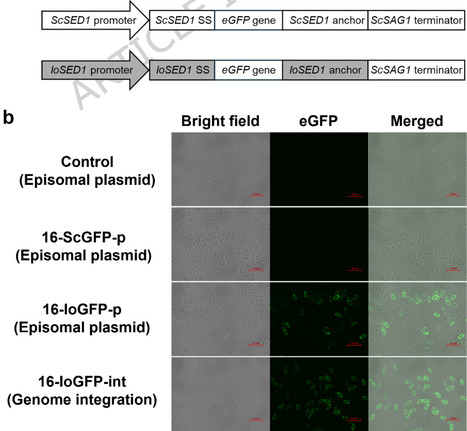
Displaying cellulolytic enzymes on the yeast cell surface via the anchoring domain of glycosylphosphatidylinositol-anchored proteins can integrate enzyme production, saccharification, and fermentation into a single step in biomass fermentation. Therefore, this technique represents a promising strategy for cost-effective and sustainable production of value-added chemicals and biofuels from lignocellulosic biomass (LCB), the most abundant renewable resource, which typically requires multiple complex processing steps. However, pretreatment of LCB, such as acidic thermochemical treatment, causes harsh cultivation conditions characterized by low pH, high temperature, and lignocellulosic fermentation inhibitors. Thus, host strains displaying cellulolytic enzymes must exhibit remarkable tolerance to these stresses. Here, we employed the non-conventional yeast Issatchenkia orientalis, which has remarkable multi-stress tolerance, and successfully established the display system in this yeast. We found that the cell-surface display cassette that functions in the model yeast Saccharomyces cerevisiae was not functional in I. orientalis; therefore, we constructed a modified display cassette based on the I. orientalis SED1 gene. Using this cassette, we successfully displayed fluorescent protein and β-glucosidase (BGL) on the cell surface. The BGL-displaying strain grew robustly on cellobiose as the sole carbon source and, notably, maintained growth even in the presence of lignocellulosic fermentation inhibitors. To the best of our knowledge, this is the first report demonstrating the successful immobilization of functional proteins on the cell surface of I. orientalis and assimilation of LCB-derived intermediates, even under stress conditions, by displaying cellulolytic enzymes, highlighting its potential as a general industrial platform for sustainable LCB bioconversion. Key points A cell-surface display cassette for Issatchenkia orientalis was constructed.The β-glucosidase-display strain utilized cellobiose as the sole carbon source.Cellobiose assimilation was observed even in the presence of fermentation inhibitors.
|
|
Scooped by
mhryu@live.com
May 19, 12:48 PM
|
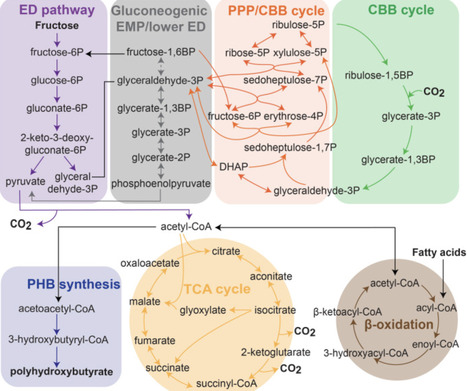
Cupriavidus necator strain H16 has emerged as a versatile microbial chassis for sustainable bioproduction due to its metabolic flexibility, enabling growth on a wide range of substrates, including H2 + CO2, formate, and organic carbon (waste) sources such as volatile fatty acids. This review first provides a comprehensive overview of recent advances in systems-level understanding and metabolic engineering of C. necator. We next discuss recent advances in omics analyses and genome-scale metabolic modeling that are increasingly used to understand the large genome and wide metabolic portfolio of C. necator. We further discuss the native metabolic network, including autotrophic growth via the Calvin Benson Bassham (CBB) cycle, as well as heterotrophic and mixotrophic capabilities. Engineering strategies to enhance substrate utilization and conversion efficiency particularly for H2 + CO2, formate, and mixed feedstocks are discussed alongside efforts to expand the substrate range in this organism. Other than the already industrialized production of the bioplastic polyhydroxybutyrate (PHB) and related polyhydroxyalkanoates in C. necator we provide an overview of the wide range of products for which proof-of-principles have been shown in C. necator. We also discuss recent advances in bioprocess design for gas fermentation, electromicrobial production, and H2-based biocatalytic reduction using C. necator. Finally, we compare C. necator with other hydrogen and formate-utilizing bacteria to identify key knowledge gaps and outline future directions for advancing C. necator as a host for sustainable industrial biotechnology.
|
|
Scooped by
mhryu@live.com
May 19, 11:57 AM
|
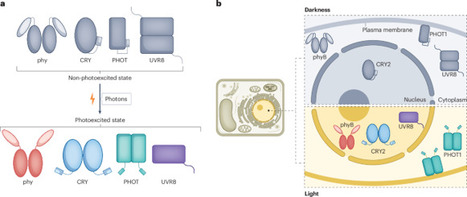
Plants decode environmental light cues through photoreceptors to orchestrate growth and developmental programs. Upon photon absorption, photoreceptor proteins transition from non-photoexcited to photoexcited states, with most functional studies focusing on the activities of the photoexcited form. However, emerging evidence reveals that non-photoexcited photoreceptors exert distinct biological functions in darkness, suggesting unexpected complexity in photon-mediated signalling. In this Perspective, we enumerate current observations underpinning photoreceptor-mediated control of plant development in darkness, categorize mechanisms of photon-dependent modulation and propose an expanded conceptual model that integrates both photoexcited and non-photoexcited activities. These insights reassess the previously overlooked importance of darkness, redefining light and darkness as dynamic, multidimensional regulators of plant physiology. In a manner distinct from their light-activated canonical roles, non-photoexcited photoreceptors can also exert biological functions in darkness, suggesting unexpected complexity in photon-mediated signalling.
|
























2st, codon optimization tool. metric: CAI: codon adaptation index; CBI: codon bias index; CUB: codon usage bias; ENC: effective number of codons; FOP: frequency of optimal codons. Relative synonymous codon usage (RSCU), Frequency of optimal codons (FOP) and more.