Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:49 AM
|
Increasing pressure from xylem-limited pathogens has driven the search for beneficial xylem-inhabiting endophytes that can enhance growth, stress tolerance, and disease resistance in woody plants. This study characterized the culturable xylem microbiota of Salicaceae species (willow and poplar) and evaluated their potential as biological control agents against vascular pathogens. A combination of microbial isolation, metabarcoding, and whole-genome sequencing was used to characterize xylem-associated bacteria. Functional traits were assessed through in vitro assays, while genome mining identified genes linked to plant-beneficial activities. Interactions between endophytes and pathogens were tested using fluorescently labeled strains in tobacco (Nicotiana tabacum) and in vitro-grown willow (Salix caprea). Bacterial genera (Bacillus, Pseudomonas, Erwinia) exhibited plant growth-promoting traits and strong antagonism against bacterial and fungal vascular pathogens, including Xylella fastidiosa, Brenneria salicis, Fusarium spp., and Verticillium dahliae. Genome analyses revealed functions related to nutrient acquisition, biofilm formation, and antimicrobial production. Co-inoculation assays significantly reduced pathogen load and disease symptoms in tobacco and mitigated symptoms in willow. Xylem endophytes act as context-dependent allies in woody plant defence. This study provides a functional and genomic framework supporting microbiome-based strategies to enhance resistance against vascular pathogens in long-lived woody hosts.
|
|
Scooped by
mhryu@live.com
Today, 1:41 AM
|
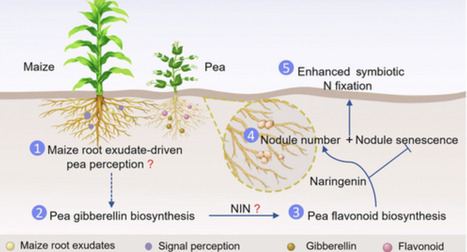
Excessive synthetic nitrogen (N) fertilizer use threatens sustainable agriculture, highlighting the need to optimize symbiotic N fixation (SNF) in cereal/legume intercropping systems. Although interspecific root interactions (IRIs) may enhance SNF, the associated regulatory mechanisms remain unclear. A 3-yr field experiment showed that Zea mays/Pisum sativum intercropping can promote SNF and N accumulation. Furthermore, glasshouse root barrier and rhizobial inoculation experiments revealed that IRIs can enhance N accumulation via pea–rhizobia symbiosis. Mechanistically, IRIs increase nodule number and inhibit nodule senescence, which are accompanied by transcriptional reprogramming and altered phytohormone abundance (e.g. gibberellin). Exogenous paclobutrazol and gibberellin treatments confirmed that SNF enhancement during IRIs involves gibberellin synthesis. Exogenous gibberellin did not affect SNF-related advantages due to IRIs, indicating that IRIs autonomously optimize endogenous gibberellin to fine-tune SNF. Genetic evidence (silencing of PsGA20OX8 and PsCHS1-4, which affect gibberellin and flavonoid biosynthesis, respectively) and pharmacological evidence (complementation with exogenous gibberellin and flavonoid, respectively) demonstrated that crosstalk between the gibberellin and flavonoid increases nodule number and inhibits nodule senescence, thereby enhancing SNF. These findings support improving legume SNF via IRIs to optimize intercropping systems and promote sustainable agriculture development.
|
|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
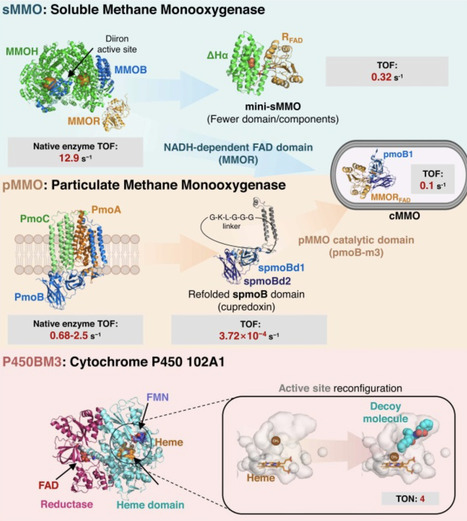
Methane (CH4) is recognized not only as a greenhouse gas but also as a promising feedstock for carbon-based chemicals. Biological methane conversion has gained attention for operating under mild conditions. Efforts have focused on enhancing methane-oxidizing enzymes, including methane monooxygenases and cytochrome P450, for both in vivo and in vitro applications. Methanol dehydrogenase is traditionally used for methanol conversion, whereas recombinant alcohol oxidase has emerged as an attractive alternative due to cofactor-independent oxidation with molecular oxygen, reduced cofactor demand, and favorable thermodynamics. Formaldehyde, produced by methanol oxidation, serves as a versatile C1 intermediate, enabling condensation reactions to form higher-value compounds. This review summarizes recent enzymatic advances, multi-step methanol upgrading pathways, and design principles, and discusses current challenges and future directions for sustainable methane valorization.
|
|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
Sequence-based predictors of recombinant protein solubility in Escherichia coli have plateaued (NESG independent-test AUC 0.760 to 0.80 over eight years of protein language-model variants). The plateau hides a latent confound: the centrifugation regime used to separate the soluble from the insoluble fraction is a hidden variable collapsed into a single binary "soluble" label. The protein's biochemistry does not change between regimes; what changes is which fraction of the lysate is recovered as soluble. Existing predictors treat the regime as label noise rather than a feature, and sequence overlap between training and test partitions masks the resulting failure mode. We release the Aiki-Sol Dataset, a tiered E. coli solubility corpus: a ~85K stringency-annotated benchmark, an Apache-licensed ~147K extension adding binary only-labelled proteins, and a ~229K research-tier pool incorporating non-commercially licensed sources. On the ~85K benchmark, scored on sequence-cluster-disjoint partitions, the strongest published binary comparator falls below chance on the 32,000 x g stratum (AUC 0.491 +- 0.020); a fine-tuned ESM-2 650M backbone with five protocol-matched outputs lifts pooled AUC by +0.108 (paired-bootstrap CI lower bound >= +0.090). The gain is curation, not architecture: structure-aware predictors given ESMFold structures do not outperform the sequence-only frame, and capacity scaled to 3B parameters does not exceed the conditioned 650M backbone. The released model, Aiki-Sol, jointly supervises five per stringency outputs alongside a marginal output for stringency-unknown proteins; on five external cohorts it lifts cohort-mean AUC from 0.69-0.70 to 0.825, with a +0.10-0.16 lift on the three cohorts at measurably-zero training-pool overlap.
|
|
Scooped by
mhryu@live.com
Today, 1:16 AM
|
Bacteroides thetaiotaomicron (Bt), a dominant bacterial species in the human gut, is a promising chassis for engineering in situ therapeutic delivery systems. Previously, we developed a secretion toolkit composed of endogenous lipoprotein signal peptides (SPs) and full-length secretory proteins from Bt. However, due to variations in length, structure, and amino-acid sequence, these SPs exhibited inconsistent secretion efficiencies across different cargo proteins. Because the activity of individual SP-cargo pairs is not readily predictable, screening and optimization is often required to achieve target secretion titers. To enhance the utility of our toolbox, we studied the impact of different SP sequence components on protein secretion, then applied this knowledge to develop a standardized toolkit to and enable predictable and tunable protein secretion across diverse SP-cargo pairs. To achieve this, we first identified the lipoprotein export sequence (LES) as the key determinant of efficient secretion of heterologous proteins by lipoprotein SPs. We next performed mutagenesis on the LES region of a representative lipoprotein SP to generate a pool of mutants featuring a standardized SP backbone with diversified LES regions. Screening and characterization of this mutant pool revealed a charge-dependent regulation of both secretion and surface display of heterologous cargo proteins. From these findings, we established a toolkit with improved tunability, enhanced predictability, and surface display capabilities that minimizes the need for iterative screening when developing protein secreting gene circuits for Bt and other Bacteroides species. By enhancing both the flexibility and control of therapeutic protein output, these results expand the potential of engineered living therapeutic applications, particularly those requiring tunable dosing or surface presentation of proteins.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
RNA-mediated control of virulence gene expression plays a crucial role in many pathogenic bacteria. However, our understanding of these processes in Streptococcus pneumoniae, a major human pathogen, remains limited. Here we discover a novel regulatory element located in the 3′-untranslated region (3′ UTR) of the mRNA encoding a major pneumococcal virulence factor, pneumococcal surface protein A (PspA). Quantitative proteomics and western blot analysis reveal that this 3′ UTR acts as a trans-acting riboregulatory element, modulating the expression of the protein chaperone, Caseinolytic protease L (ClpL), in a temperature-dependent manner. We show that it is the full-length pspA mRNA, and not the pspA-3′-located processed F5 RNA, which is involved in the regulation of ClpL expression, with the sRNA-interacting exoribonuclease Cbf1 playing an important role. Furthermore, complement deposition assays show that the regulatory pspA-3′ UTR contributes to inhibition of complement C3 deposition in a PspA-independent and temperature-dependent manner. This discovery adds a new dimension to our understanding of PspA’s role in bacterial virulence, highlighting an intricate layer of RNA-mediated regulation that contributes to the pathogenicity of S. pneumoniae.
|
|
Scooped by
mhryu@live.com
Today, 12:19 AM
|
Upstream open reading frames (uORFs) in the 5′ leader of bacterial mRNAs can modulate gene expression, yet genome-wide identification remains limited. We combined bioinformatic prediction of ribosome-binding sites (RBS), a Shine Dalgarno sequence and a start codon, with experimental validation to uncover new uORFs in Sinorhizobium meliloti 2011. From totally 1106 predicted upstream RBSs (uRBSs), we first examined 15 candidates using eGFP reporters and integrating existing RNA-seq and Ribo-seq data. Translation was detected at 13 sites, with fluorescence intensity broadly correlating with predicted initiation rates. Two uRBSs correspond to gene start sites, thereby refining gene annotations. In nine cases, uRBS mutations affected downstream gene expression in reporter fusions. Among others, the data suggests that a Type I secretion system operon, the RNA chaperone gene hfq, and metabolic genes are regulated by uORFs. Four uORFs acted through translational coupling. We also identified uRBSs that were ribosome-occupied yet (nearly) silent in eGFP assays, and closely spaced to the downstream main RBS (mRBS). These uRBSs probably mediate ribosomal occlusion downregulating lacR and SM2011_RS36230. A re-screen of the prediction set revealed 335 close uRBS/mRBS pairs. Three of them were analyzed, supporting the proposed ribosomal occlusion mechanism for SM2011_RS03630 and SM2011_RS22110, while for glnK translational coupling to an uORF was suggested. These results indicate that uORFs are more widespread in bacteria than previously recognized and suggest that direct ribosomal occlusion of the mRBS is a novel mechanism for down regulating protein synthesis.
|
|
Scooped by
mhryu@live.com
May 14, 11:34 PM
|
Surfactin production by Bacillus subtilis is typically performed under aerobic conditions, requiring high aeration and agitation, which leads to mechanical stress for the cells and therefore promotes excessive foaming. As a process-oriented alternative to full aerobic operation, an aerobic to micro-aerobic switching strategy was developed that aims to decouple biomass formation from surfactin synthesis by controlling oxygen availability. Promoter activities relevant to nitrate respiration and surfactin biosynthesis were first characterised in shake flasks using transcriptional reporter strains and online monitoring of dissolved oxygen. The nitrite reductase promoter showed the strongest induction under oxygen-limited conditions and was used to construct an oxygen-responsive production strain in which the native PsrfA promoter was replaced to suppress surfactin formation during aerobic growth and shift production to the micro-aerobic phase. The switching concept was subsequently evaluated in 30-L stirred-tank bioreactor cultivations using stepwise reductions of dissolved oxygen setpoints combined with exponential glucose feeding and nitrate supplementation. The engineered strain B. subtilis MG19 enabled stable transitions into micro-aerobic operation without apparent loss of biomass, while a reference strain B. subtilis MG17 with the native regulation showed surfactin formation already during the aerobic phase and pronounced process disturbance after switching, characterised by glucose accumulation and a strong decrease in surfactin concentration. Overall, the study demonstrates oxygen switching as a scalable process engineering tool for controlling surfactin production phases and highlights key constraints for robust micro-aerobic bioreactor operation.
|
|
Scooped by
mhryu@live.com
May 14, 11:26 PM
|
Faithful DNA replication, which is a highly orchestrated process, is essential in all living organisms to ensure accurate transmission of genetic information to their descendants. In this review, we summarize the molecular mechanisms and dynamics of DNA replication in E. coli and compare them with those of the phylogenetically distant B. subtilis. Although the central features of replication initiation, elongation, termination, and restart are broadly conserved, distinct mechanisms have evolved to adapt each bacterium to its complex environment. This review highlights the players and outlines both the shared and divergent molecular mechanisms governing how the multi-component replication machines, the replisomes, are assembled at the origin of chromosomal replication, oriC, on a circular genome, undergo bidirectional replication elongation, and are ultimately disassembled upon reaching the terminus of replication, ter, region. In response to stress, (a) replication restart mechanism(s) operating at sites other than oriC re-assemble the replisome, allowing unidirectional DNA synthesis to resume, thereby ensuring completion of the cell cycle and maintenance cell viability.
|
|
Scooped by
mhryu@live.com
May 14, 11:16 PM
|
As a renewable energy source, biofuels can reduce dependence on fossil fuels and effectively lower greenhouse gas emissions, mitigating the global climate change issue. The production of liquid biofuels primarily relies on the biosynthetic processes of microorganisms. While many natural strains possess specific metabolic capabilities to produce liquid biofuels, their performance often fails to meet the demands of industrial-scale production. Therefore, genetic engineering to construct strains with optimized metabolic pathways has become an inevitable approach. Synthetic biology technology has advanced rapidly in recent years, giving rise to a large number of new tools and methods that significantly accelerate the development of strain-engineering, thereby enhancing the yield and productivity of biofuels. In this article, we review the latest advancements in the synthetic biology method of developing liquid biofuel-producing strains. We summarize the progress in synthetic biology from the perspectives of chassis strain selection, the development of synthetic biology tools, and metabolic regulation tools for strains. Additionally, by showcasing examples of strain development for some important biofuels, we demonstrate the immense potential of synthetic biology in biofuel production. Through these summaries, this article provides valuable references for the development of biofuel-producing strains, contributing to the further advancement and application of biofuel technologies.
|
|
Scooped by
mhryu@live.com
May 14, 4:37 PM
|
The trait‑based root economics space provides a fundamental framework for understanding plant belowground adaptation to environmental change, yet it typically omits root exudation that sustains plant resource acquisition and health. Results from a ~3500 km north‑south forest transect demonstrate that arbuscular mycorrhizal (AM) trees have higher root exudation rates than ectomycorrhizal (ECM) trees, with stronger increases under warmer and wetter climates. Root exudation rate is positively related to specific root length and negatively related to root diameter for both mycorrhizal types, but is related to tissue density and nitrogen content only in AM trees. These findings suggest that root exudation may represent an alternative collaboration strategy, facilitating organic nutrient acquisition more strongly for AM species and functioning as a relatively redundant nutrient acquisition mechanism for ECM species. Moreover, mycorrhizal type modulates how exudation rates relate to the “fast–slow” strategy. Overall, our findings refine the conceptual framework of the root economics space and enhance understanding of fine‑root strategies. Trees with different mycorrhizal types show distinct root exudation rates and trait associations. Here the authors report that mycorrhizal type modifies the position of root exudation within the root economics space, revealing contrasting nutrient acquisition strategies.
|
|
Scooped by
mhryu@live.com
May 14, 3:39 PM
|
Bacterial viruses can send and receive signals to inform critical decisions. In this issue of Cell, Manley et al. and Gallego-del-Sol et al. independently identify crosstalk between bacterial viruses, leading us to wonder what the information warfare landscape between them looks like.
|
|
Scooped by
mhryu@live.com
May 14, 1:32 PM
|
This century, global human health has been marked by a seemingly increasing list of outbreaks and epidemics caused by RNA viruses — masters of rapid evolution, host switching and immune escape. With a propensity for mutation, coupled with recombination, reassortment and extensive population interactions, RNA viruses generate remarkable genetic diversity within constrained evolutionary landscapes. Although most mutations are deleterious, a subset fuels adaptation to selective pressures and environments, potentially enabling pathogens to reach new hosts and become epidemic and pandemic threats. Recent advances in molecular virology have clarified how mutation biases, genome organization, epistasis and host factors shape viral diversity, revealing both vulnerabilities and evolutionary constraints. These mechanisms underlying viral evolution are now being leveraged to design evolution-informed countermeasures. These include live-attenuated vaccines with reduced risk of reversion, antivirals that target mutationally constrained regions or drive populations towards extinction, or universal vaccines directed against conserved regions. Looking forward, the integration of high-throughput mutational mapping, structural biology and computational modelling, including artificial intelligence-driven predictive tools, promises to transform our ability to anticipate viral evolutionary trajectories. This Review discusses how embedding evolutionary principles into translational virology may improve preparedness for future outbreaks by shifting the field from reactive to predictive strategies. In this Review, Tran and Vignuzzi examine how molecular processes and evolutionary pressures shape RNA virus diversity and adaptation. They outline how genome structure, mutational pathways and viral interactions influence evolutionary outcomes, offering insights relevant to the design of better vaccines and antiviral strategies.
|
|
|
Scooped by
mhryu@live.com
Today, 1:45 AM
|
Arsenic contamination threatens rice (Oryza sativa) production, yet the synergistic use of iron plaque (IP) and root-associated biofilms as a rhizosphere barrier to limit arsenic uptake remains unexplored. To address this, we engineered an arsenic-resistant (AR) plant growth-promoting rhizobacterium (AR-PGPR), Bacillus subtilis p43-Taglo1, expressing the speciation-inert arsenic-binding protein TaGlo1. In a contaminated paddy, this strain increased grain yield by 10.7–11.6% and reduced grain arsenic by 28.2–37.4% compared to the wild-type. The engineered strain robustly colonized roots and enhanced the formation of a functional IP-biofilm composite, which sequestered more arsenic. This was driven by a 2.87-fold increase in Fe(II) oxidation and elevated production of extracellular polymeric substances (EPS) (1.4-fold) and siderophores (1.5-fold). Transcriptomic analysis revealed that inoculation upregulated bacterial genes for Fe(II) oxidation, siderophore, and EPS biosynthesis, while in rice roots, it activated phytohormone pathways and downregulated arsenite transporters (OsLsi1 and OsLsi2). We conclude that AR-PGPR can restore beneficial root–microbe interactions under arsenic stress. The IP-biofilm composite acts as an inducible barrier essential for the dual benefits of arsenic exclusion and growth promotion. Our study shows that AR rhizobacteria fortify the IP-biofilm composite to reduce arsenic uptake and promote rice growth, providing a route toward safer rice production in arsenic-affected regions.
|
|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
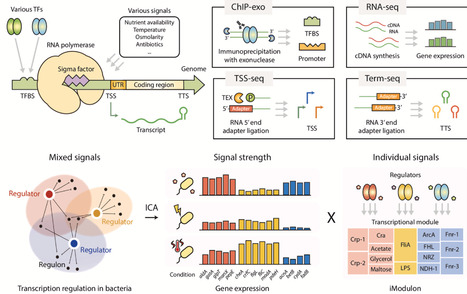
Advances in next-generation sequencing (NGS) have provided refined insight into bacterial transcriptional regulatory networks (TRNs) on a genome-wide scale. The integration of genomic and transcriptomic datasets has clarified the architecture of TRNs, revealing regulatory complexity that requires computational methods for interpretation. The TRNs and their regulatory elements characterized through these analyses serve as a foundation for systems biology. In this review, we discuss the progression from targeted molecular characterization to systems-level approaches. NGS-based characterization at near-single-base-pair resolution has allowed precise interrogation of DNA-binding protein dynamics. Machine learning-based frameworks, such as independent component analysis, have enabled the identification of co-regulated gene modules and the discovery of novel regulatory relationships within transcriptomic datasets. In addition, deep learning models have shown utility in uncovering transcriptional regulatory elements and guiding the de novo design of regulatory sequences. Together, these tools are being integrated into closed-loop biofoundry platforms, accelerating automated workflows in bacterial systems engineering.
|
|
Scooped by
mhryu@live.com
Today, 1:29 AM
|
Mechanistic understanding of gut ecology is limited by the availability of tools for precise manipulation of microbiome composition. Here, we isolate lytic phages to enable targeted removal of gut commensal Escherichia fergusonii (Ef) from complex, undefined stool-derived in vitro communities. A single phage drove resistance without fitness cost in monoculture, but resistant Ef exhibited reduced fitness in communities, enabling expansion of closely related Proteobacteria. Resistance arose via reversible promoter inversion linked to outer-membrane function. A phage cocktail overcame resistance to achieve Ef knockout across communities with minimal collateral effects. Using knockout communities, we show that Ef is necessary and sufficient for preventing Salmonella invasion. Replacement with an Ef transposon-mutant library revealed that community-specific fitness defects are enriched in genes involved in outer-membrane assembly. Disruption of these genes sensitized Ef to antagonistic community members, highlighting interspecies warfare as a key driver of microbiome ecology. These results establish phage-mediated perturbation as a framework for linking species to community-level function and for enabling precision microbiome engineering.
|
|
Scooped by
mhryu@live.com
Today, 1:19 AM
|
Bacteriophage receptor-binding proteins (RBPs) determine bacterial host recognition and are central to phage host-range evolution, yet the structural principles governing how RBPs diversify and expand host range remain not fully understood. Although phage RBPs are thought to evolve through modular exchange of receptor-binding domains, it is unclear how this modularity is organized across diverse RBP architectures, at what structural scales recombination operates, and how it shapes host-range breadth. Here we combined large-scale AlphaFold3 structural modelling, domain-level annotation, de novo pseudo-domain segmentation, sequence modularity analysis and experimentally determined host-range phenotypes across 192 Klebsiella phages and 382 high-confidence RBPs spanning up to 96 K-types. We generated the first system-wide structural atlas of Klebsiella phage RBPs, resolving 39 structurally distinct RBP-classes. Although capsule-degrading beta-helix depolymerases dominated numerically, 161 RBPs across 37 RBP-classes employed non-depolymerase architectures, including 18 novel RBP-classes. Structural and sequence analyses show that this diversity arose through modular reuse of structural domains rather than independent invention. Conserved N-terminal scaffolds linked depolymerase and non-depolymerase RBPs across morphotypes, while receptor-binding regions diversified through recombination operating at and beyond domain boundaries. We found recent modular exchange both within genera, where capsule-specific and capsule-independent RBPs can be swapped to alter host-range strategy, and across morphotypes, where depolymerase modules moved between distant lineages altering capsule specificity. Together, these architectures resolve into six receptor-recognition strategies, establishing multi-scale modularity as the primary organising principle of RBP diversification and a structure-informed framework for guiding phage isolation and engineering against Klbsiella pneumoniae. The complete atlas is freely accessible as an interactive community resource at Klebsiella-Phage-RBP-Atlas.
|
|
Scooped by
mhryu@live.com
Today, 12:57 AM
|
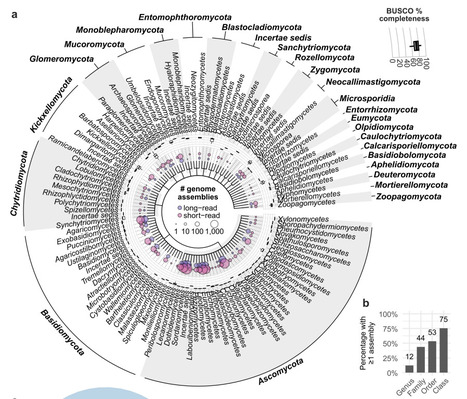
Fungal genomics has expanded rapidly over the past 30 years, and recently the pace and breath has further quickened for many taxa, although many taxonomic gaps persist. With three decades of rapid growth, fungal genomics now merits a re‑examination of its history, progress, and unresolved taxonomic gaps. Here, we review the development of fungal genomics from early efforts such as the Fungal Genome Initiative to current progress driven by third-generation long-read sequencing. We have compiled and summarised publicly available fungal genomes to highlight trends in assembly quality, adoption of long-read technologies, and taxonomic representation. Notably, substantial phylogenetic gaps remain, particularly outside Dikarya, and significant challenges persist for unculturable taxa. This review identifies priorities for the fungal community, including: (1) coordinated efforts to close major taxonomic gaps across the fungal tree of life; (2) improved repository metrics to facilitate identification of high-quality assemblies; and (3) improved and standardised genome annotation which is lacking for most assemblies. Together, these steps will support the development of reliable genomic resources that capture the full breadth of diversity across the fungal kingdom, generating foundational data for comparative genomics, evolutionary biology, functional studies, genetic studies and applied research.
|
|
Scooped by
mhryu@live.com
Today, 12:24 AM
|
Membrane transport is a fundamental biological process with profound implications for pharmacology, biotechnology, and microbiology. While computational approaches have largely adopted a protein-centric perspective to annotate transportomes, inferring transport function directly from the intrinsic properties of substrates remains a major challenge. Addressing transport at the compound level enables the systematic evaluation of whether molecules undergo active transport and by which mechanisms, independent of prior transporter annotation. Here, we introduce ChemProFlow, a comprehensive computational framework that redefines transport analysis from a substrate-centric perspective. By integrating geometric deep learning with orthology-based genomic mapping, ChemProFlow predicts molecular transportability, assigns transport mechanisms according to the Transporter Classification Database, and identifies the microorganisms encoding the corresponding transport systems. We show that this integrated pipeline enables scalable, end-to-end mapping of substrate-transporter-organism relationships, with broad applications in pharmacology for anticipating drug transport, in biotechnology for guiding strain engineering, and in microbiology for dissecting substrate utilization across diverse taxa. By capturing the chemical determinants of transportability, ChemProFlow generalizes to previously unseen substrates and provides a high-throughput framework for systematic exploration of molecular transport across diverse biological contexts.
|
|
Scooped by
mhryu@live.com
Today, 12:11 AM
|
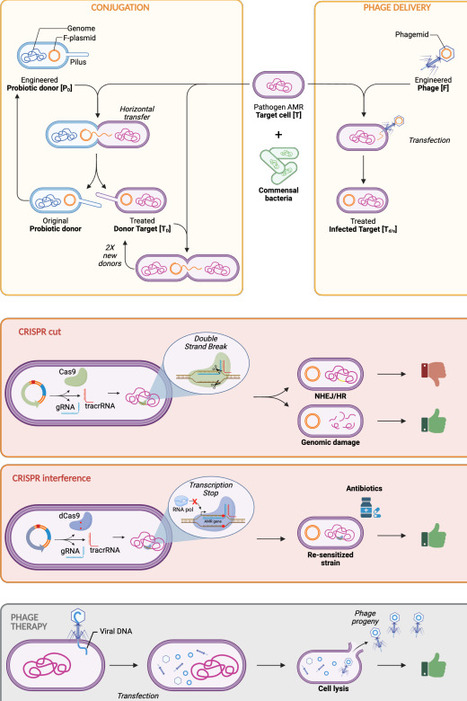
Antimicrobial resistance (AMR) poses a significant threat to global health by diminishing the effectiveness of conventional antibiotics. This study aims to assess, using a systems biology approach, a potential synthetic biology-based strategy that employs engineered conjugative probiotic bacteria and bacteriophages to combat AMR, examining the implications of implementing targeted gene silencing as an alternative to direct bacterial killing. A comprehensive mathematical model was developed to describe the dynamics of the delivery systems (engineered conjugative probiotic bacteria and engineered phages) and the antimicrobial actuators being studied (genome cutting via CRISPR systems and AMR-gene silencing through CRISPR interference), also compared to traditional phage therapy (selection of phages capable of killing pathogens through bacterial-specific viral infection). The target population includes antibiotic-resistant bacteria competing with other probiotic bacteria for colonizing the host environment. The model explicitly incorporates parameters for mutations that affect actuator functionality and simulates their impact on overall therapeutic performance. Simulations show how variations in actuator efficiency and emergence of new mutations in target pathogens affect the long-term suppression of resistance genes. Including mutational effects provides insights into system robustness and guides optimal therapeutic design choices. The proposed modeling framework effectively captures key biological and mechanistic aspects of engineered therapies, enabling the prediction and optimization of each intervention against resistant pathogens. It highlights engineered phages and CRISPR interference as the most promising candidates for the design of new engineered biological therapeutics. This study establishes a quantitative foundation for rational design and dosage optimization in engineered phage- and bacterial-based therapies, advancing the use of synthetic biology methods to fight antimicrobial resistance.
|
|
Scooped by
mhryu@live.com
May 14, 11:30 PM
|
Sustainable biomanufacturing requires moving beyond fossil and agricultural carbon sources. To date, industrial biotechnology depends largely on starch, sugar, and plant-derived glycerol, creating competition with food and feed production and pressure on limited arable land. Yet, microbial metabolism is not inherently constrained to these substrates, opening opportunities for alternative feedstocks. Non-agricultural carbon sources, particularly single- and two-carbon (C1 and C2) compounds, offer a compelling alternative. Produced from CO2 via electrochemical or biological routes, methanol, formate, acetate, and ethanol connect renewable energy with microbial synthesis while enabling carbon recycling. Their use, however, introduces distinct metabolic and thermodynamic constraints, including limitations in energy conservation, redox balance, and pathway driving forces. Here, we examine C1 and C2 assimilation pathways in yeasts, highlighting key bottlenecks and engineering advances that make sustainable circular biomanufacturing possible.
|
|
Scooped by
mhryu@live.com
May 14, 11:21 PM
|
Artificial trans-encoded small RNAs (atsRNAs), which repress translation by base pairing with target mRNAs, have proven to be powerful tools for regulation of gene expression for reconstructing cell factories and genetic circuits. Currently, rationally designed atsRNAs use mostly E. coli natural trans-acting sRNAs with an Hfq-binding structure and a Rho-independent terminator as scaffolds. However, owing to the structural differences in Hfq proteins between E. coli and other bacteria, the inhibitory efficacy of atsRNAs may be influenced by heterologous sRNA scaffolds, highlighting the necessity of screening native sRNA scaffolds in various bacteria to develop functional synthetic sRNAs. The root-associated bacterium Pseudomonas stutzeri A1501 is a well-studied strain regarding nitrogen fixation, yet post-transcriptional regulatory platforms for this species remain scarce. In this study, we describe the development of an atsRNA platform by employing the natural Hfq-dependent sRNA CrcZ scaffold from the nitrogen-fixing strain P. stutzeri A1501, which is conserved in the Pseudomonas strain. On the basis of the CrcZ scaffold, two atsRNAs targeting the master nitrogen-fixing regulatory complex encoded by the nifLA operon were developed and found to effectively suppress nifLA gene expression at the posttranscriptional level. Further studies demonstrated that the 5′ untranslated region of target mRNAs and five Hfq-binding sites are optimal features for designing atsRNAs using CrcZ as a scaffold. The well-established effective design principle for synthetic sRNAs with the CrcZ sRNA scaffold will enable more rational and efficient engineering of synthetic gene networks in Pseudomonas strains.
|
|
Scooped by
mhryu@live.com
May 14, 4:43 PM
|
Codon usage bias has a crucial impact on the translation efficiency and cotranslational folding of proteins, necessitating the algorithmic development of codon optimization/harmonization methods, particularly for heterologous recombinant protein expression. Codon harmonization is especially valuable for proteins sensitive to translation rates because it can potentially replicate native translation speeds, preserving proper folding and maintaining protein activity. This work proposes a Monte Carlo-based codon harmonization algorithm, MoSAiC (Monte Carlo-based Simulated Annealing for Linked Codons), for the harmonization of a set of linked codons, which differs from conventional codon harmonization by focusing on the codon sets rather than individual ones. Our MoSAiC demonstrates robust computational performance on ribosomal proteins (S18, S15, S10, and L11) as model systems. Among them, the harmonized gene of RP S18 was expressed and compared with the expression of the wild-type gene. The harmonized gene clearly yielded a larger quantity of the protein, from which the amount of the soluble protein was also significant. These results underscored the potential of the linked codon harmonization approach to enhance the expression and functionality of sensitive proteins, setting the stage for more efficient production of recombinant proteins in various biotechnological and pharmaceutical applications.
|
|
Scooped by
mhryu@live.com
May 14, 4:34 PM
|
Sequence-based deep learning has advanced genome interpretation, yet most models remain task-specific and rely on retraining, limiting scalability across biological contexts. Here we present SUCCEED, a supervised multi-task DNA foundation model pretrained on 6,389 ENCODE functional genomics tracks to learn transferable regulatory representations. By integrating convolutional layers with a Transformer architecture, SUCCEED captures both local sequence motifs and long-range regulatory dependencies, achieving performance comparable to or exceeding Enformer across benchmark tasks. Through transfer learning, it predicts cell-type-specific epigenomic profiles, denoises sparse chromatin accessibility signals, and predicts three-dimensional chromatin contacts without CTCF input across data scales and cell types. Across diverse genomics tasks, SUCCEED performs comparably to supervised foundation models such as Sei and outperforms self-supervised models trained solely on DNA sequence. Overall, SUCCEED is a transferable and scalable foundation model that provides a unified framework for genome-scale regulatory modeling in complex biological contexts. SUCCEED is a supervised multi-task DNA foundation model pretrained on 6,389 ENCODE tracks. It integrates CNN and Transformer architectures to model local motifs and long-range dependencies, enabling prediction of genomic regulatory features across tasks and cell types.
|
|
Scooped by
mhryu@live.com
May 14, 1:43 PM
|
The molecular drivers and fitness trade-offs controlling the transition of environmental bacteria into human pathogens remain enigmatic. Here, we describe a small RNA (sRNA) with a unique modular structure that shapes the evolution of toxigenic Vibrio cholerae, the agent of cholera. The sRNA comprises a variable 5’ module located within the ompU ORF and a conserved 3’ region downstream. This atypical location confers a distinct bimodular structure to the OmpU-encoded sRNA (OueS), generating allelic variants with unique functions. Unlike environmental counterparts, the OueS allele from toxigenic strains controls phenotypes essential during host colonization in a domain-dependent manner: a) inhibits biofilm formation by suppressing RyhB, b) confers resistance against intestinal bacteriophages by activating the CBASS system, and c) drives mucus penetration. Toxigenic OueS is essential for successful intestinal colonization and acts as a functional surrogate of the master virulence regulator ToxR, controlling over 84% of its regulome. However, this allele leads to reduced competitive fitness during colonization of natural reservoirs when compared to environmental ones. Finally, we determined that the acquisition of the ompU ORF is modular and controlled by exogenous conditions, providing critical insights into the evolution of toxigenic V. cholerae and the emergence of pathogenic traits in bacteria. In this work, authors describe a modular small RNA that drives the emergence of virulence traits in the cholera pathogen. It controls key colonization phenotypes, acting as the functional surrogate of a major virulence regulator, and is associated with environmental fitness trade-offs.
|