 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 9:30 AM
|
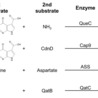
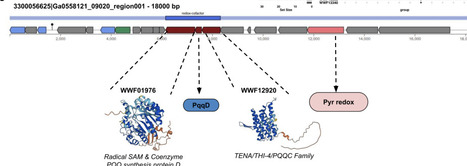
QatABCD is a widespread antiphage defense system in prokaryotes comprising four protein components. QatC, a signature component, is homologous to QueC, an enzyme involved in nucleobase modification during queuosine biosynthesis. QatA and QatD are predicted to function as an ATPase and a nuclease, respectively, while QatB lacks identifiable sequence motifs. Here, we report the structural and functional characterization of QatB and QatC. We determine the structure of QatC bound to the ATP analog AMPPNP and perform structure-guided functional assays. We further find that QatB and QatC form a stable heterodimer and solve the structure of the QatB–QatC complex. In addition to determining the structure of QatB, structural analysis suggests that it may serve as a substrate of QatC, implicating a potential regulatory mechanism. These findings provide structural and functional insights into QatB and QatC, laying a foundation for understanding the molecular mechanism of the QatABCD system in phage defense. QatABCD is a widespread prokaryotic anti-phage defense system. Here, authors solved the crystal structures of AMPPNP-bound QatC and QatB-QatC homodimer complex, suggesting that QatB may serve as a substrate of QatC.
|
|
Scooped by
mhryu@live.com
Today, 1:33 AM
|
A significant body of research has been devoted to pinpointing and cataloguing the binding sites of RNA-binding proteins (RBPs) on target transcripts. The most common techniques involve crosslinking and immunoprecipitation (CLIP) followed by high-throughput sequencing. In this review, we provide a comprehensive summary of the major advancements in CLIP-based techniques and state-of-the-art data analysis methods designed for identifying and analyzing the binding sites of RBPs. We also brief on methods used to determine the functional relevance of these binding sites and, in addition, delve into the major hurdles faced in the detection and elucidation of the binding sites of RBPs. Finally, we explore reproducibility concerns in the CLIP field, and conclude by suggesting potential avenues for future improvements.
|
|
Scooped by
mhryu@live.com
May 13, 11:13 PM
|
All folded proteins continuously fluctuate between their low-energy native structures and higher-energy conformations that can be partially or fully unfolded. These rare states influence protein function, interactions, aggregation and immunogenicity, yet they remain far less understood than protein native states. Although native protein structures are now often predictable with impressive accuracy, conformational fluctuations and their energies remain largely invisible and unpredictable, and experimental challenges have prevented large-scale measurements that could improve machine learning and physics-based modelling. Here we introduce a multiplexed experimental approach to analyse the energies of conformational fluctuations for hundreds of protein domains in parallel using intact protein hydrogen–deuterium exchange mass spectrometry. We analysed 5,778 domains 28–64 amino acids in length, revealing hidden variation in conformational fluctuations, even between sequences sharing the same fold and global folding stability. Site-resolved hydrogen exchange nuclear magnetic resonance analysis of 13 domains showed that these fluctuations often involve entire secondary structural elements with lower stability than the overall fold. Computational modelling of our domains identified structural features that correlated with the experimentally observed fluctuations, enabling us to design mutations that stabilized low-stability structural segments. Our dataset enables new machine-learning-based analysis of protein energy landscapes, and our experimental approach promises to profile these landscapes at considerable scale. An analysis of 5,778 domains 28–64 amino acids in length reveals hidden variation in conformational fluctuations, even between sequences sharing the same fold and global folding stability.
|
|
Scooped by
mhryu@live.com
May 13, 4:12 PM
|
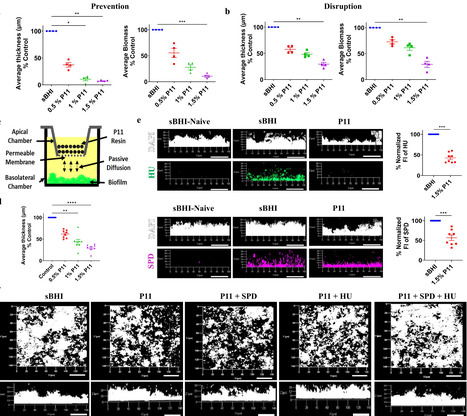
Biofilms are structured multicellular bacterial communities encased within an extracellular matrix comprised of exopolysaccharides, proteins, extracellular DNA (eDNA), and other biopolymers that provide protection against environmental stressors. We and others have shown that eDNA serves as a fundamental structural element common to even multispecies biofilms. During biofilm maturation, ubiquitous DNABII proteins facilitate the conversion and stabilization of eDNA into the rare and rigid Z-DNA conformation, thereby enhancing matrix integrity and rendering the underlying eDNA resistant to nucleases. We have previously shown that the removal of positively charged molecules, such as DNABII proteins, results in rapid, significant disruption of diverse biofilms. Here, we identify the polyamine spermidine as another essential positively charged molecule that, together with DNABII proteins, contributes to the development and maintenance of the eDNA-dependent extracellular matrix. We also provide evidence that SPD is present within the biofilm matrix alongside DNABII proteins in multiple bacterial pathogens. Our findings indicate that SPD and DNABII proteins cooperate to promote Z-DNA formation. Depletion of SPD and DNABII using cation exchanger P11-phosphocellulose or inhibition of SPD synthesis via dicyclohexylamine impaired biofilm formation and destabilized preformed biofilms. These results suggest that polyamine synthesis or accumulation represents a potential target for biofilm disruption and control.
|
|
Scooped by
mhryu@live.com
May 13, 3:27 PM
|
Autonomous microrobots can reach hard-to-access regions in the human body for minimally invasive therapy. However, their microscale size limits the integration of on-board memory, making their operation dependent on external controls. Here, we develop a magnetic probiotic microrobot integrated with memory-capable genetic circuit to execute autonomous antitumor treatment. Through a one-time magnetic hyperthermia trigger, the biological thermal sensor in the microrobot perceives temperature change and activates the memory module Bxb1-ssrA-attB-P7-attP, transferring the microrobots into a therapeutic state to continuously degrade fibrin and soften the tumor microenvironment. The genetic memory remains active for at least 12 days. A synergy toward deep tumor penetration is subsequently established between the memory-encoded softening and the physical penetration through magnetically controlled wave-like locomotion of microrobots. Compared with memory-absent microrobots, the proposed microrobots achieve a 6.70-fold tumor matrix stiffness reduction and boost in vivo anticancer efficacy from 21.86 to 87.52%. Beyond oncology, the proposed system establishes a generalizable framework of memory-encoded medical microrobots.
|
|
Scooped by
mhryu@live.com
May 13, 3:02 PM
|
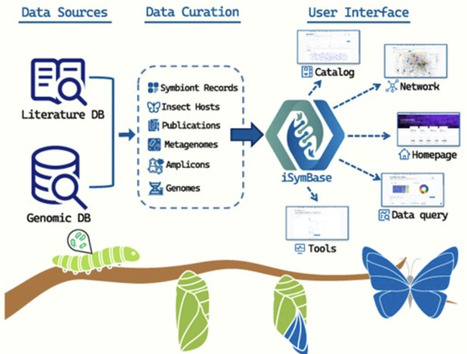
Insect symbionts play essential roles in host biology, influencing nutrition, immunity, reproduction, and environmental adaptation, ultimately shaping insect physiology, ecology, and evolution. With the rapid growth of functional and genomic datasets on insect symbionts, there remains a critical need for a dedicated platform to systematically compile, organize, and analyze these datasets from an integrative ecological perspective. Here, we developed an insect Symbiont database, named as iSymBase, by manually curating functional records and genomic datasets of insect symbionts from published academic literature. Currently, iSymBase contains over 2,657 insect symbiont functional records spanning 795 host species, along with 1,494 metagenomes, 14,992 amplicon datasets, and standardized genome and gene catalogs, providing a comprehensive resource for ecological and comparative insect symbiont researches. iSymBase offers standardized query functionalities, such as data browsing, keyword associative search, sequence alignment, data download and submission. Beyond conventional database functionalities, iSymBase provides several innovative tools: insect-symbiont interaction network for host-symbiont ecological relationships, a batch annotation tool for detecting ecologically functional symbionts from microbiome profiles, and an AI-powered chatbot iSymSeek designed to assist researchers with related knowledge queries. Taken together, iSymBase will serve as an open-access and continually updated platform for storing, querying, and analyzing insect symbiont data, supporting ecological exploration of host–symbiont interactions, symbiont functional diversity, and microbiome-driven adaptation.
|
|
Scooped by
mhryu@live.com
May 13, 2:54 PM
|
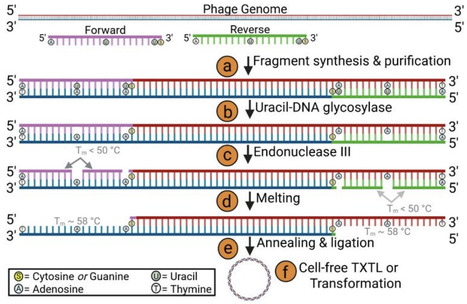
Synthetic biology enables the creation of systems such as bacteriophage (phage)-based biosensors, leveraging the innate specificity and efficiency of phages to rapidly identify pathogens. However, the current genome assembly and editing methods, including Gibson Assembly, Golden Gate Assembly, and CRISPR-Cas systems, have limitations that can hinder speed and flexibility, especially when complex modifications are needed. This study introduces a novel means for generating engineered bacteriophages through a one-pot, modular in vitro genome assembly platform utilizing uracil-DNA glycosylase, which allows genome modification without requiring extended overlaps, the removal of restriction enzyme sites, a Cas system, or homologous recombination. The design also minimizes the risk of secondary structure formation (e.g., hairpins), allowing for a more efficient assembly of fragments. To demonstrate functional genome engineering, we incorporated a NanoLuc luciferase reporter gene into the T7 genome, producing a recombinant phage capable of detecting E. coli, a strategy consistent with our previous work on waterborne pathogen detection. This platform enables rapid and flexible synthetic genome construction with high functional assembly efficiency, with broad applications in phage engineering, biosensing, and synthetic biology.
|
|
Scooped by
mhryu@live.com
May 13, 2:42 PM
|
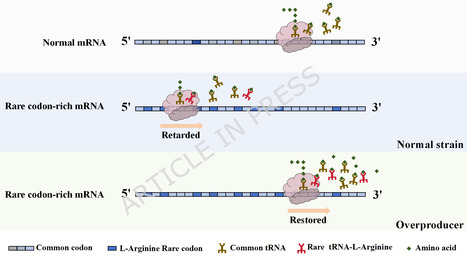
Rapid advancements in complex engineered metabolic system design have not been matched by corresponding screening and identification capabilities for high-performing microbial variants, creating considerable pacing constraints in strain development. Therefore, advancing high-throughput phenotyping methods is essential for propelling synthetic biology and metabolic engineering towards scalable biomanufacturing. Accordingly, we developed a rare codon-dependent fluorescent biosensor that enables real-time, high-content monitoring of intracellular L-arginine accumulation. This system employed L-arginine-rich peptide modules that are engineered with AGG rare codons fused to the StayGold fluorescent protein, developing a stringent link between fluorescence intensity and cytoplasmic L-arginine levels. By integrating ultraviolet mutagenesis with fluorescence-activated cell FACS sorting, we efficiently isolated superior producers from an engineered Escherichia coli ARG library, achieving a screening efficiency of 55.12%. The top-performing isolate, E. coli ARG-B10, exhibited a 94.8% enhancement in L-arginine production. Its plasmid-cured derivative, E. coli B10, was used for scale-up fermentation, attaining a 120.5 g/L titer and 0.45 g/g glucose yield under industrially relevant conditions. Genomic analysis revealed missense mutations in key metabolic genes (coaBC, gst, yihU, and fruB), indicating improvements in the precursor supply and redox management. This biosensor platform operates independently of orthogonal translation systems and is readily applicable to wild-type strains, offering a powerful and generalizable tool for accelerating microbial strain optimization.
|
|
Scooped by
mhryu@live.com
May 13, 2:03 PM
|
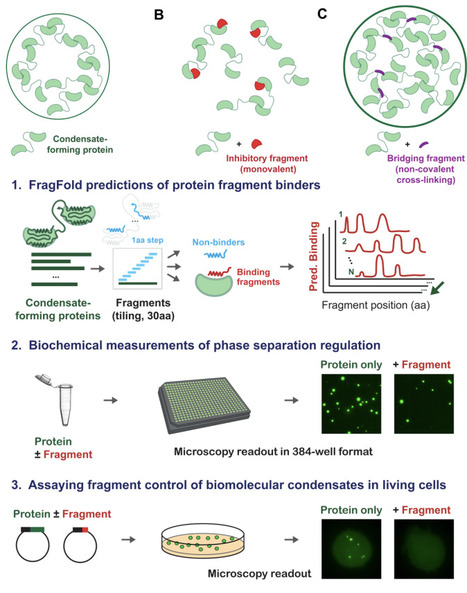
Biomolecular condensates are a major driver of cellular organization; however, we lack a predictable and systematic approach to modulate the multivalent interactions underlying their formation. Here, we demonstrate that the AI-driven FragFold method enables robust and generalizable design of protein fragments to control biomolecular condensate formation. We apply this approach across diverse proteins: G3BP1, SARS-CoV-2 nucleocapsid, TDP-43, and focal adhesion kinase (FAK). Computationally screening 2,235 fragments, we selected 18 candidates for further investigation. Overall, we attain a 50% success rate (9/18 designs) in discovering condensate-controlling protein fragments, experimentally testing just 3-5 candidates per protein. For each condensate-forming protein, the success rate is at least 40%. Furthermore, FragFold-predicted fragment binding modes align with their condensate-inhibitory or enhancing activities, revealing both known and newly identified interactions underlying condensate formation. In FAK, a condensate-inhibitory fragment uncovered a domain interaction required for phase separation, and mutational analysis validated its importance. Notably, this inhibitory fragment also suppresses FAK condensate formation in living mammalian cells. Together, these results establish AI-guided protein fragment discovery as a generalizable strategy to dissect and control the molecular interactions that govern biomolecular condensates.
|
|
Scooped by
mhryu@live.com
May 13, 1:19 AM
|
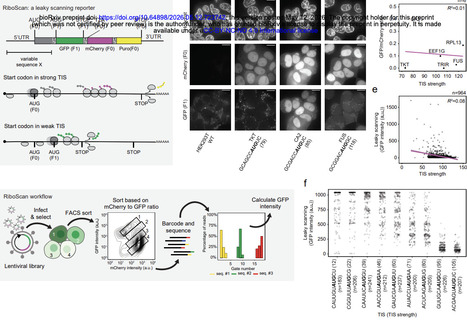
Accurate selection of start codons by ribosomes is a fundamental determinant of proteome composition. Although the 'Kozak sequence'—an 8-nucleotide sequence flanking the start codon—has long been viewed as the primary determinant of initiation in eukaryotes, it fails to explain the large diversity of start codon usage across transcripts. Here we combine massively parallel reporter assays, bioinformatics, machine learning, single-molecule imaging and cryo-electron microscopy to define the 'extended translation initiation sequence (eTIS)', an ~80-nucleotide sequence surrounding the start codon that governs initiation efficiency. A deep-learning model trained on eTIS features accurately predicts translation initiation across transcripts. Unexpectedly, we find that the Kozak sequence is not optimal for initiation as is widely presumed, and we identify the origin of this discrepancy. eTIS nucleotides that promote efficient initiation are enriched in the human transcriptome and are evolutionarily conserved, underscoring their functional importance. Biophysical and structural analyses reveal that specific eTIS residues—including the key +6 position and residues in the mRNA entry and exit channel—engage ribosomal proteins, rRNA and initiation factors to promote start codon recognition by stabilizing the ribosome at the start codon and facilitating the structural transitions required for initiation. Finally, optimization of the eTIS markedly enhances translational fidelity and protein output from therapeutic mRNAs, highlighting its practical utility. Together, these findings redefine the sequence logic of translation initiation and establish a framework for precise control of protein expression.
|
|
Scooped by
mhryu@live.com
May 13, 12:28 AM
|
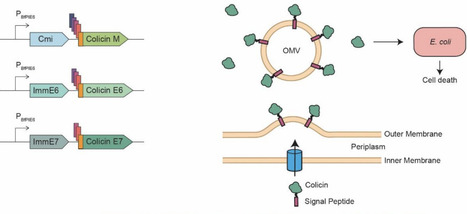
Urinary tract infections (UTIs) are among the most common bacterial infections globally and create a large burden on the healthcare system. Uropathogenic Escherichia coli (UPEC) account for the majority of UTIs and increase the risk of recurrence. The standard treatment is antibiotics and, with the rise of multi-drug resistant UPEC lineages, there is a need for alternative treatments and prevention. Colicins, bacteriocins targeting and produced by E. coli, have previously been shown to inhibit the growth of pathogenic E. coli and are a promising alternative. Here, we engineer commensal Bacteroidaceae to secrete colicins via outer membrane vesicle (OMV) targeting signal peptides to suppress E. coli in the mouse gut. Secreted colicins were assessed for their ability to kill primary clinical isolate UPEC strains, including epidemic multi-drug resistant ST131 strains, along with other pathogenic and type strains. Specifically, secreted colicin E7, from Phocaeicola vulgatus fully eliminated of several UPEC strains in culture. In mice, P. vulgatus secreting colicin E7 prevented the extended colonization of two clinical UPEC strains and restored microbiome diversity. Together, this work shows the viability of secreted, heterologous antimicrobials from P. vulgatus as prophylactic treatment against the colonization of pathogenic E. coli utilizing cross-phylum antagonism in the gut.
|
|
Scooped by
mhryu@live.com
May 12, 11:54 PM
|
Wastewater surveillance has emerged as a critical tool for global epidemiology, yet the functional diversity of wastewater microbiomes remains poorly characterized at the protein level. Here, we present WasteFams, the first comprehensive database dedicated to the systematic exploration of protein families in wastewater metagenomic and metatranscriptomic studies worldwide. Integrating data from 580 metagenomes, 132 metatranscriptomes, and 1,709 reference genomes, WasteFams catalogs 3,887 non-redundant protein families (containing ⪰100 members) derived from over 105 million predicted proteins. Each protein family is enriched with multi-layered annotations, including AlphaFold3 structural predictions, taxonomic classifications, and biome-specific metadata. To further expand their functional annotation, we integrated deep genomic context analysis to link protein families to Mobile Genetic Elements (MGEs), Biosynthetic Gene Clusters (BGCs), Antibiotic Resistance Genes (ARGs), and CRISPR elements. Accessible through the EnvoFams portal, WasteFams provides a user-friendly interface featuring advanced search capabilities, sequence and structural similarity tools, and interactive visualization modules. As global initiatives increasingly leverage wastewater for public health and environmental insights, WasteFams can serve as a critical resource for discovering novel microbial functions, monitoring resistance mechanisms, and exploring the biotechnological potential of secondary metabolites within wastewater-engineered ecosystems.
|
|
Scooped by
mhryu@live.com
May 12, 11:50 PM
|
Alkaloids represent a large and structurally diverse class of natural products predominantly found in plants and rarely in animals. Well-known compounds such as vinblastine, berberine, and scopolamine exhibit remarkable pharmaceutical potential, with several already in clinical use. These plant-derived alkaloids have attracted enduring interest due to their diversity, structural complexity, and pronounced biological activities, making them a privileged resource for drug discovery. In recent years, breakthrough advances have been made in elucidating the biosynthetic pathways of plant alkaloids, making this a highly promising research field. Successful cases including reserpine, strychnine, and hyoscyamine have not only provided novel strategies for drug development and sustainable production, but also greatly stimulated scientific enthusiasm. In this review, we focus on representative plant alkaloids from major classes including monoterpene indole alkaloids (MIAs), tetrahydroisoquinoline alkaloids (THIQAs), tropane alkaloids (TAs), and other types, covering the period from 2018 to 2025. It highlights key challenges in pathway elucidation, including stereochemical control (R/S configuration), important cyclization, and tailoring modifications particularly those catalyzed by specific enzymes. This review also covers catalytic sequence determination, and innovative approaches for long-pathway decoding. Furthermore, we discuss how synthetic biology and metabolic engineering strategies enable efficient and sustainable microbial production of these compounds. By identifying common obstacles and proposing effective solutions, this review aims to inspire researchers engaged in functional gene characterization, synthetic biosystems development, yield optimization, and drug innovation for plant alkaloids. It seeks to promote interdisciplinary collaboration across botany, chemistry, biology, and pharmaceutical sciences, thereby accelerating the discovery and scalable production of high-value plant alkaloids.
|
|
|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
In recent years, the development of peptide drugs has seen significant growth. These molecules often go beyond simple linear chains composed of the standard 20 amino acids. Peptide drugs frequently incorporate non-standard amino acids, non-amino components, and can exhibit mono- or multicyclic structures, branching, and other complex topologies. Consequently, there is a growing need for accessible tools that allow researchers to easily generate and modify 1D, 2D, and 3D representations of these complex peptides, serving as a starting point for further optimization. PEP-EDIT was created to meet this need. It offers a user-friendly, interactive web interface for generating complex peptide representations from 1D BILN (Boehringer Ingelheim Line Notation) sequences, using a customizable monomer library. Building on the pyPept library, PEP-EDIT enhances its functionality with options such as pH-dependent protonation and simplified specification of conformational constraints. The platform leverages interactive 2D and 3D visualizations to guide peptide design, offers intuitive management of monomers and 3D models, and includes collaborative and interactive visualization tools. PEP-EDIT is available at https://pep-edit.rpbs.univ-paris-diderot.fr. This website is free and open to all users and there is no login requirement.
|
|
Scooped by
mhryu@live.com
May 13, 11:45 PM
|
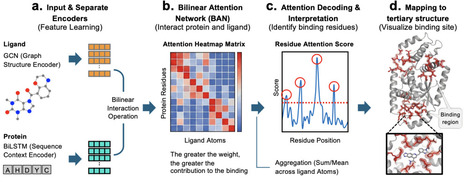
Rapid and accurate prediction of protein-ligand bindings is essential for drug discovery. While generative AI has driven rapid advancements in structure-based approaches, sequence-based methods remain significantly faster and more cost-effective. Here, we present a weakly supervised deep learning framework integrating graph convolutional networks (GCN) for molecular encoding and bidirectional long short-term memory (BiLSTM) for protein modeling. The latter represents long-range dependencies better than the widely used convolutional neural network (CNN). Leveraging a bilinear attention network (BAN), this model learns protein-ligand pairwise interactions without requiring three-dimensional structural supervision. By using the publicly available BindingDB dataset, the model was trained, solely on affinity labels, and successfully classified binder and non-binders with AUROC of 0.96 and an AUPRC of 0.95. The model generates interpretable attention maps that serve as a "GPS" to locate binding sites. Remarkably, despite the lack of structural training data, it can pinpoint key contact residues confirmed by crystal structures. Our method could function as a scalable filter for giga-scale libraries, allowing rapid screening of drug candidates with direct structural insights into the protein-ligand interface.
|
|
Scooped by
mhryu@live.com
May 13, 11:09 PM
|
Single cell protein (SCP) is a promising alternative to animal-derived protein, offering high productivity with low environmental impact. However, SCP performance varies widely across microbial species. This review systematically and quantitatively compares 57 bacterial SCP candidates in terms of safety, protein quality, and production efficiency, integrating multivariate analysis to highlight representative high-performing species and remaining data gaps, and providing a framework for rational species selection toward sustainable protein supply.
|
|
Scooped by
mhryu@live.com
May 13, 3:56 PM
|
Obesity is a heterogeneous condition comprising a continuum of phenotypes with various metabolic and inflammatory profiles. Metabolically healthy obesity (MHO) identifies individuals with obesity but a relatively preserved metabolic state, although little is known about the gut microbiome features underlying this phenotype. Here, we analyzed gut microbial network structures of 931 individuals living with metabolically healthy non-obesity (MHNO), MHO, metabolically unhealthy non-obesity (MUNO), and metabolically unhealthy obesity (MUO), performing cross-sectional analyses on feces shotgun metagenomics data. Individuals with MHNO and MHO harbor more robust and functionally cohesive microbial networks, while communities from MUO and MUNO phenotypes exhibit a potentially dysbiotic state with reduced connectivity. A nutritional intervention cohort showed an improvement in network connectivity in parallel with metabolic improvements. Our findings show differences in microbial connectivity and association patterns across metabolic and obesity phenotypes, shedding light on how distinct microbial network structures may associate with host metabolic health and disease. Here, the authors show that microbiome network topology associates with metabolic health across obesity phenotypes, with reduced connectivity in metabolically unhealthy states and improved network structure following a nutritional intervention.
|
|
Scooped by
mhryu@live.com
May 13, 3:08 PM
|
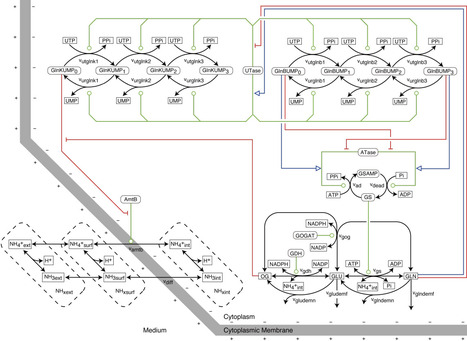
Nitrogen is essential for all life forms, and microorganisms prefer ammonium as a nitrogen source. Due to the low affinity of glutamine synthetase (GS) for ammonium, E. coli must maintain high intracellular ammonium (NH4+) concentrations to sustain its rapid growth. Under ammonium limitation, E. coli imports ammonium through the transporter AmtB and incorporates it into glutamine by using GS. On the basis of structural and mutagenesis information, mechanisms have been proposed for the transport of ammonia (NH3) and protons by AmtB through spatially (partly) separate routes. These mechanisms do not explain the required coupling between proton and ammonia transports. How does the membrane potential push the ammonia inward so as to attain high concentrations near GS? We here compare six candidate kinetic models of E. coli ammonium transport and assimilation in terms of how they reproduce experimental data from the literature: three variants of the 'electro-binding model' in which the membrane potential affects AmtB-NH4+ binding, and three variants of the 'electro-flipping model' in which it influences the conformational flip of the transporter. The computer simulations decide that the electro-binding models are 28 times more plausible than the electro-flipping models and suggest that the transmembrane electric potential affects AmtB-NH4+ binding from the cytoplasmic side. The addition of kinetic and thermodynamic features to existing structural information plus our requirement of an explanation of the coupling, suggest a new spatiotemporal mechanism of coupling of ammonia and proton flows in AmtB. Further simulations show that GS and AmtB regulation is coordinated via both the uridylyltransferase/uridylyl-removing enzyme (UTase) and 2-oxoglutarate binding, allowing the cell to minimize futile cycling while maintaining rapid growth. The free energy cost of transport-related futile cycling exceeded that of the GS reaction itself. Moreover, AmtB enabled robust growth under varying ammonium concentrations and pH levels, albeit at a cost of futile cycling that became substantial at low ammonium. These findings highlight the crucial roles of GS and AmtB in E. coli's adaptations and provide new insights into the trade-off mechanism between nutrient acquisition and energy efficiency.
|
|
Scooped by
mhryu@live.com
May 13, 2:58 PM
|
E. coli Nissle 1917 (EcN) is a promising chassis strain in synthetic biology, but its application is limited by inefficient genetic manipulation. This study established an efficient genetic manipulation system for EcN via electroporation optimization and recombinase-mediated cassette exchange (RMCE). Systematic screening revealed that using SB medium for cultivation and sterile deionized water as the wash buffer significantly improved EcN electroporation efficiency. Further optimization of electroporation conditions enhanced RMCE efficiency. We constructed the recombinant strain EcN-lox by inserting the loxP-hyg-lox5171 cassette into the EcN genome (replacing the colibactin-synthesizing gene clbB), which served as a stable landing pad for site-specific integration of exogenous genes via RMCE. Compared with direct electroporation, this RMCE system exhibited superior efficiency in integrating large exogenous DNA fragments, successfully mediating the integration of 17 kb and 29 kb gene cluster segments, while direct electroporation failed to stably maintain large plasmids in wild-type EcN. Finally, the RMCE system was applied to integrate a 10-kb artificial astaxanthin biosynthetic operon into EcN, achieving successful heterologous astaxanthin production. The highest yield (0.627 mg g−1 DCW) was obtained when the recombinant strain was cultured in LB medium at 37 °C for 24 h in shake flasks. Collectively, the optimized electroporation protocol and RMCE-mediated genome integration system developed in this study provide valuable tools for EcN genetic engineering, facilitating its applications in heterologous production of valuable natural products and other synthetic biology fields.
|
|
Scooped by
mhryu@live.com
May 13, 2:46 PM
|
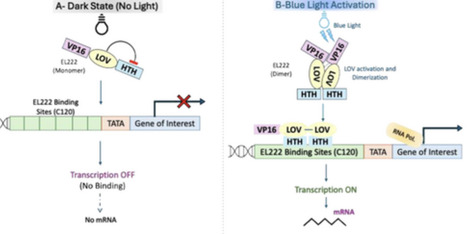
Methylotrophic yeasts such as Pichia pastoris are widely used for heterologous protein production because they contain strong and tightly regulated promoters. However, the use of methanol as an inducer presents several practical challenges, including toxicity, flammability, high oxygen demand during fermentation, and increased production costs. To overcome these limitations, researchers have been working on redesigning the AOX1 regulatory system and developing alternative induction strategies that do not rely on methanol. One promising approach is optogenetics, which uses light to control gene expression in a non-invasive way. These systems rely on light-sensitive proteins such as phytochromes, cryptochromes, LOV-domain proteins, and UVR8, allowing gene activity to be regulated in a precise and reversible manner without adding chemical inducers to the culture medium. This review brings together key advances in yeast optogenetics, with a focus on the EL222 system, highlighting its implementation for light-controlled heterologous protein production in P. pastoris and its broad application in synthetic biology and metabolic engineering in Saccharomyces cerevisiae. The growing versatility and scalability of EL222-based circuits highlight their potential to reshape both fundamental research and industrial bioprocessing through safer, more controllable, and energy-efficient gene regulation strategies.
|
|
Scooped by
mhryu@live.com
May 13, 2:37 PM
|
Reinforcement learning (RL) has been used to control a wide range of dynamic processes, especially ones that are too complex to model well or have stochastic environmental perturbations. Fed-batch fermentations are subject to changes in starting cell growth rates and process variations that can affect cell growth and secreted target production. RL has been shown on digital environments of fermentation to control known setpoints (such as temperature) but has yet to be demonstrated for unconstrained product maximization. In this work we develop a fed-batch fermentation model (digital twin) of Aspergillus niger secreting glucoamylase using the Monod model, known literature parameters, and assumed constants to align with typical production values. An RL agent is trained on this environment to evaluate types of algorithms (Proximal Policy Optimization (PPO) and Soft Actor-Critic (SAC)), rate of learning, and effects of process perturbations. State variables fed to the model include run time, cell concentration, and measured enzyme activity in the fermentation broth, with the objective of maximizing the enzyme activity. It is found that SAC outperforms PPO, achieving 77.7% of the maximum quality with 200 training episodes and 95% at the 2400th episode, compared to PPO which achieves 80% of the max reward after 2912 episodes of training. The RL controller is benchmarked against a traditional, model-free controller that used Bayesian optimization to discover the optimal feed rate for a given cell type. The traditional controller can be implemented with fewer training runs; however, it is not as robust when exposed to variations in starting cell growth or process perturbations including faulty feed or cooling pumps. In all cases, the RL controller can maintain higher enzyme production, despite changes in the process. Finally, the RL controller is exposed to new cell types (in silico) to determine the experimental cost of updating the trained model with real bioreactor runs. Surprisingly, we found that with no updates the model can perform well across a wide range of new cell types, and that by retraining the quality of performance improves. These results indicate that an in silico trained RL agent can be updated with an array of fermentation experiments to provide robust fermentation control.
|
|
Scooped by
mhryu@live.com
May 13, 1:32 AM
|
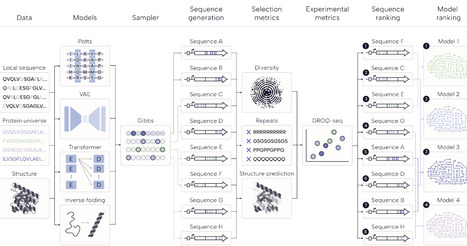
Generative models are increasingly used for protein design, but the lack of standardized evaluation frameworks limits comparison across model classes and hinders translation to experimental success. Here, we introduce a unified sampling and benchmarking framework that enables controlled sequence generation across alignment, protein language, and structure-based models, and apply it to Tobacco etch virus (TEV) protease. Across hundreds of thousands of designed sequences, different models explore distinct regions of sequence space with no clear computational selection metrics to assess enzymatic function. Experimental evaluation reveals large differences in functional outcomes, ranging from non-functional variants to sequences with ~9-fold higher activity than wildtype. Machine learning-designed libraries achieve a 39.32% hit rate (percentage of variants matching or exceeding wildtype activity) compared to 6.06% for an error-prone PCR baseline. Structure-based models perform best overall, with hit rates of 74.4% and 66.8% for ESM-IF1 and ProteinMPNN, respectively. Commonly used selection metrics do not strongly correlate with experimental activity, highlighting a gap between in silico evaluation and enzyme function. Together, these results establish a generalizable framework for benchmarking generative protein models and demonstrate the necessity of experimental validation for guiding model development and sequence prioritization.
|
|
Scooped by
mhryu@live.com
May 13, 1:07 AM
|
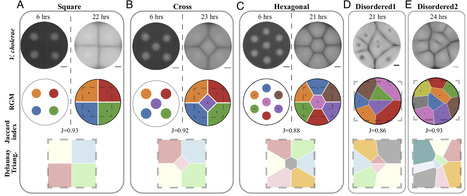
The organization of bacteria has a central role in shaping interactions, dynamics, and composition within communities and microbiomes. Bacteria form distinct spatial patterns that have often been attributed to microbial processes such as chemotaxis, nutrient transport, and signaling. However, common patterns are observed across distinct bacteria and conditions, suggesting that a general organizing principle could direct bacterial organization. Here, we find that the organization of bacteria is explained by geometric ordering that promotes space-filling efficiency, giving rise to geometric patterns known as Voronoi tessellations. We find that the Voronoi Growth Model accurately predicts bacterial pattern formation in diverse conditions including in biofilms at the liquid–air interface, swimming populations, the zebrafish gut, and conditions that promote swarming. The patterns are observed in two and three dimensions, at the cm and mm length scales, across diverse species (Vibrio cholerae, Pseudomonas aeruginosa, E. coli), arise solely from the principles of Voronoi tessellation, and require no detailed knowledge of microbial processes. Entropic considerations show that bacteria provide little or no information about the pattern formation, which is determined solely by their initial positions and environmental conditions. These findings demonstrate that bacterial communities achieve robust, reproducible organization through a universal geometric principle, linking microbial patterning to the broader biological context of multicellular organization.
|
|
Scooped by
mhryu@live.com
May 13, 12:03 AM
|
Wastewater surveillance has emerged as a critical tool for global epidemiology, yet the functional diversity of wastewater microbiomes remains poorly characterized at the protein level. Here, we present WasteFams, the first comprehensive database dedicated to the systematic exploration of protein families in wastewater metagenomic and metatranscriptomic studies worldwide. Integrating data from 580 metagenomes, 132 metatranscriptomes, and 1,709 reference genomes, WasteFams catalogs 3,887 non-redundant protein families (containing ⪰100 members) derived from over 105 million predicted proteins. Each protein family is enriched with multi-layered annotations, including AlphaFold3 structural predictions, taxonomic classifications, and biome-specific metadata. To further expand their functional annotation, we integrated deep genomic context analysis to link protein families to Mobile Genetic Elements (MGEs), Biosynthetic Gene Clusters (BGCs), Antibiotic Resistance Genes (ARGs), and CRISPR elements. Accessible through the EnvoFams portal, WasteFams provides a user-friendly interface featuring advanced search capabilities, sequence and structural similarity tools, and interactive visualization modules. As global initiatives increasingly leverage wastewater for public health and environmental insights, WasteFams can serve as a critical resource for discovering novel microbial functions, monitoring resistance mechanisms, and exploring the biotechnological potential of secondary metabolites within wastewater-engineered ecosystems.
|
|
Scooped by
mhryu@live.com
May 12, 11:52 PM
|
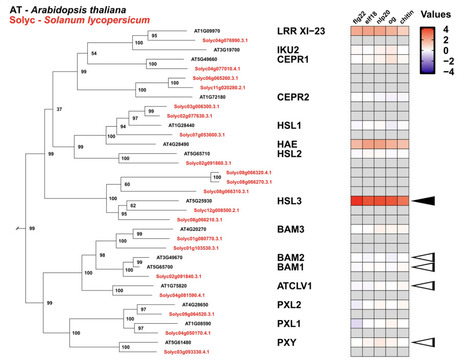
Gene family functional exploration often requires analyzing motifs, domains, and associated datasets (e.g. gene expression) in the phylogenetic context of a gene tree. As genomic resources become more abundant, local pipelines are needed to analyze gene families of interest with project-specific resources. Here we present BLAST-Align-Tree (BAT), a bioinformatic pipeline for automated gene family phylogeny construction and annotation to enable gene tree exploration. BAT combines a BLAST search of local genome databases with a robust and flexible gene tree construction pipeline that enables multiple modes of annotation. Output visualizations display experimental datasets, custom regex specified amino acid motifs, and protein HMM domain annotations. For flexibility, BAT runs locally and is independent of pre-existing databases, allowing the easy incorporation of custom genomes and datasets. Three primary case studies described here demonstrate the utility of BAT for inferring the function of homologs and orthologs within characterized gene families. BAT is suitable for fine scale phylogenomic analysis of gene families across the tree of life, and default genomes available on installation span model eukaryotes.
|




















r-2st