 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
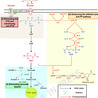
Shikimic acid is a crucial precursor for the production of indole derivatives and chiral drugs, and the biomanufacturing offers reduced cost and simplified process compared to traditional plant extraction. Current research on industrial-scale production of shikimic acid achieves notable progress, yet the limitations associated with high production cost, complex operation process, and excessive byproduct formation still persist. To tackle these difficulties, this study aimed at establishing a high-efficient shikimic acid-producer through systematic modification and fermentation optimization in Escherichia coli. First, the route for the synthesis of shikimic acid was divided into three modules (i.e., sugar uptake, bioproduct synthesis, and metabolite catabolism), of which the disruption of glucose uptake system, reinforcement of bioproduct synthesis, and block of its catabolism rewired metabolic fluxes and provided the possibility for efficient synthesis. Next, series of systematic engineering involving in deleting pyruvate bypass, reinforcing the pentose phosphate (PP) pathway, and adjusting promoter strength promoted precursor supply and avoided metabolic overflow. The best strain EC022 generated 5.27 g/L of shikimic acid, highlighting the significance of precursor availability. Then, fermentation conditions including substrate concentration, inoculation size, cultivation temperature, induction time, and C/N ratio were comprehensively optimized, which boosted the bioproduction to 8.70 g/L in the optimized conditions. Finally, a final titer of 61.87 g/L shikimic acid was achieved in a 7 L bioreactor. This study developed an efficient shikimic acid-producer with industrial potential through metabolic engineering and bioprocess optimization.
|
|
Scooped by
mhryu@live.com
Today, 12:53 AM
|
E. coli couples the initiation of DNA replication with cell size by modulating the activity of the replication initiator protein DnaA. The activity of DnaA is regulated by both its interconversion between an active and inactive form and its titration on binding sites on the chromosome. Whereas its interconversion has been thoroughly studied, the extent to which DnaA titration can control replication initiation is poorly understood. Here, we describe the control of E. coli DNA replication via titration by modulating the expression of an 'always-active' DnaA variant in four growth conditions. While we obtained stable cell cycles during slow growth, faster growth associated with overlapping replication forks led to replicative instability and DNA damage. Overall, our results provide insights into the limits of titration-based systems in the control of genome replication and their potential role in the evolutionary trajectory of E. coli. Finally, this study provides design principles for a simplified, titration-only regulatory mechanism for DNA replication in synthetic cells.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
Photosynthetic microorganisms can convert sunlight and CO₂ directly into biomass and bioproducts. Yet, most biofoundries still optimize heterotrophic chassis reliant on agricultural sugars, limiting impact on global decarbonization. This review argues that sustainable manufacturing requires integrating microalgae and cyanobacteria into Design–Build–Test–Learn pipelines and shifting from biomass conversion to light- and CO₂-driven production. We highlight advances in genetic and cellular engineering in model photosynthetic microbes, including modular cloning, genome editing, and organelle engineering that enable pathway design for lipids, isoprenoids, and proteins. We discuss phototroph-specific bottlenecks for automation and standardization, including slower growth, variable transgene expression, chlorophyll autofluorescence, and the need for controlled illumination and gas exchange with linked data pipelines. Finally, we examine cultivation and scale-up constraints, emphasizing co-optimization of strain traits, reactor design, and downstream processing to improve techno-economic and environmental performance. Photosynthetic biofoundries are therefore both necessary and increasingly feasible for a low-carbon bioeconomy.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
We showed previously that yeasts in Starmerella and Wickerhamiella genera (W/S clade) exhibit numbers of horizontal gene transfer (HGT) events much larger than found in any other yeast species, with bacteria and other fungi (Pezizomycotina) as donors. Here we shed light on HGT in the W/S clade by carrying out the characterization of xenologous genes present in three species harboring together close to 600 xenologous genes. Metabolic genes were strongly overrepresented and either introduced functions new to yeasts or restored functions that were likely absent in the W/S clade ancestor. RNA-sequencing revealed lower global levels of expression of xenologous vs native yeast genes in two species. This difference is associated with the preferential accumulation of xenologous genes in large, AT-enriched chromosome terminal regions, dubbed “End” domains, characterized by overall lower gene expression. In one species, “End” domains were shown to be more permissive for protein diversification, but only for xenologous genes. We posit that “End” domains may generate favorable conditions for the adaptation and retention of xenologous genes, helping explain the exceptional numbers of HGT events in the W/S clade. Horizontal gene transfer is generally considered uncommon in eukaryotes, but rates are very high in a floral yeast clade. Underlying this may be large, low-expression, chromosomal safe harbors that promote retention and adaptation of incoming genes.
|
|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
Sialic acids cap the ends of many glycan chains and play pivotal roles in cell signaling, immunity, and pathogen interactions. However, the fast synthesis of sialylated glycans by automated glycan assembly (AGA) has remained a long-standing challenge. Here we show a general strategy that leverages macrobicyclic sialic acid building blocks to achieve reliable α(2,3)- and α(2,6)-sialylation on solid support. Using this method, a collection of nine sialylated human milk oligosaccharides (HMOs) is assembled, including fucosyldisialyllacto-N-tetraose (DSLNF II), a highly branched, fucosylated structure that is very difficult to synthesize by solution-phase methods. An improved global deprotection protocol provides access to pure, functionalized complex glycans suitable for further biological studies. This work provides the broadly applicable solution for automated chemical sialylation, opening the door to prepare collections of sialylated glycans for biomedical research. Human milk oligosaccharides (HMOs) are essential components of breast milk and about one-fifth of all known HMOs contain sialic acids, which have key functional roles. Authors show a strategy for the rapid synthesis of sialylated glycans using automated glycan assembly (AGA) to create nine complex HMOs, including highly branched structures.
|
|
Scooped by
mhryu@live.com
May 9, 11:58 PM
|
Microbial death pathways (MDPs) are increasingly recognized as key drivers of terrestrial carbon cycling, primarily through their regulation of microbial necromass carbon (MNC), a critical pool in global carbon dynamics. Yet explicit representation of MDPs in soil organic carbon (SOC) models remains limited. Here, we developed and evaluated three SOC models that differ in their structure of MNC pool: the multiple-pathway necromass (MPN) model, which partitions microbial necromass carbon (MNC) into four MDP-derived subpools; the dual necromass (DUN) model, which differentiates two necromass pools with distinct decay rates; and the single necromass (SIN) model, which aggregates necromass into a single pool. Using a unified data assimilation framework and SOC observations from six major agricultural regions in China, we found that the MPN model consistently outperformed DUN and SIN models across most regions, producing necromass subpool dynamics, scenario responses, and parameter sensitivities that closely reflect the mechanistic understanding of MDPs. In cold or nutrient-limited regions, however, the simpler DUN model performed similarly while requiring fewer parameters, emphasizing the importance of balancing model complexity with regional ecological constraints. Our results demonstrate that explicitly incorporating MDPs enhances the robustness and mechanistic realism of SOC simulations and provides a robust foundation for more explicit representations of MDPs to assess the soil carbon sequestration potential and guide sustainable land management.
|
|
Scooped by
mhryu@live.com
May 9, 11:44 PM
|
Diatoms are promising microorganisms to provide sustainable routes for photosynthetic terpenoid production from CO2, yet their potential for compartmentalized engineering remains largely unexplored. Here, we systematically profiled the biosynthetic capacity of Phaeodactylum tricornutum by targeting representative synthases for hemi-, mono-, sesqui-, and tetraterpenoids to the cytosol, chloroplast, and periplastidial compartment (PPC). This comprehensive analysis revealed that all major prenyl phosphate precursors, DMAPP, GPP, FPP, and GGPP, are accessible in all compartments, including in the PPC, and can sustain heterologous flux without major physiological penalties, although production efficiency varies across compartments and product classes. By determining precursor availability, we propose the diatom PPC as an engineerable intracellular space directly integrated with a eukaryotic complex chloroplast, establishing a foundation for further compartmentalized terpenoid biosynthesis engineering strategies. Moreover, we highlight its utility as a unique interface to investigate the metabolic exchange between the MVA and MEP pathways. These findings provide a systematic framework for compartment-specific terpenoid engineering in diatoms and open new opportunities for modular pathway assembly and synthetic biology in photosynthetic eukaryotes.
|
|
Scooped by
mhryu@live.com
May 9, 11:17 PM
|
Metagenomic workflows involve complex multi-step analyses, from quality control and assembly to binning, annotation, and strain-level profiling. Few existing metagenomic pipelines achieve the combination of flexibility, reproducibility, and hybrid assembly support within a unified workflow. We present StrainMake, a Snakemake-based workflow for de novo metagenomic analysis from short, long, or hybrid sequencing data. StrainMake integrates widely used tools across all major steps—quality control, assembly, binning, dereplication, taxonomic and functional annotation—while also providing non-redundant gene catalogues, community-scale metabolic models, and strain-level microdiversity metrics. The modular design enables the use of alternative tools, scalable execution on HPC systems, and full reproducibility through Snakemake and Conda. Applied to the CAMI II strain-madness dataset, StrainMake produced high-quality assemblies and metagenome-assembled genomes (MAGs), while enabling strain-resolved comparisons across samples. Hybrid assemblies improved contiguity, whereas short-read assemblies offered faster runtimes, illustrating the workflow’s benchmarking capacity.
|
|
Scooped by
mhryu@live.com
May 9, 5:15 PM
|
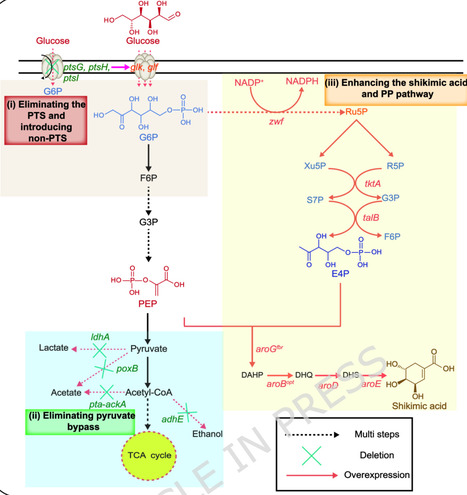
Brown rot wood-degrading fungi release carbon (C) from deadwood but leave behind a large fraction of C sequestered in lignin residues or as fungal metabolites. The strength of sequestration in these C residuals remains unclear, but proteobacteria-dominated bacterial communities have been implicated in metabolizing C from decay residues, possibly erasing the C sequestration potential assumed for brown rot. Here, we paired a model brown rot fungus (Rhodonia placenta) with a model Alphaproteobacterium (Rhodopseudomonas palustris) to track fungal release and bacterial utilization of C derived from decaying wood. We found that fungal decay products generated by R. placenta could be used by R. palustris for growth, and later decay stages contained more usable substrates than early stages. High performance liquid chromatography with mass spectrometry identified a range of aromatic and non-aromatic compounds in the fungal-decayed wood, but after 95 days of bacterial growth, R. palustris preferentially consumed non-aromatic acids over aromatic lignin monomers. Genes involved with aromatic compound degradation were unimportant for bacterial growth, and RNA sequencing revealed that aromatic compound degradation genes were repressed on decayed wood extract. Randomly barcoded transposon sequencing failed to identify a solitary catabolic pathway used by R. palustris, suggestive of substrate co-utilization, and surprisingly, showed that genes involved with copper toxicity were essential. Finally, we found that genes involved with biosynthesis of certain cofactors and amino acids were no longer essential on decayed wood extract, suggesting these nutrients were readily accessible. This study helps lay the foundation to understand potential bacterial-fungal interactions in decayed wood.
|
|
Scooped by
mhryu@live.com
May 9, 5:02 PM
|
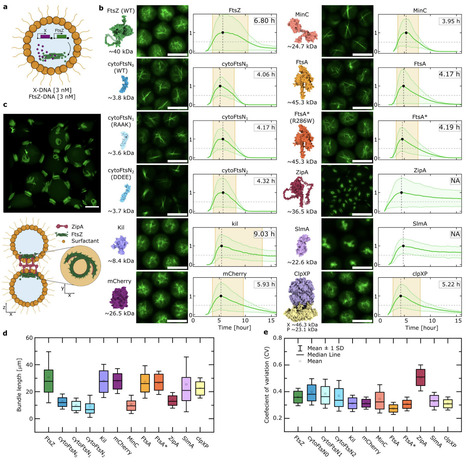
Designing minimal biological systems with emergent functions such as spatiotemporal self-organization is a central goal of bottom-up synthetic biology. While computational optimization and design show promises to accelerate functional protein engineering through Design-Build-Test-Learn cycles, screening libraries for complex functions remains a major challenge. Conventional screens typically lack the spatiotemporal resolution and cell-like confinement required in bottom-up synthetic biology. Here, we present PUREdrop, an automated microfluidic platform that encapsulates and expresses protein libraries in thousands of picolitre-sized synthetic cells per construct. These droplets are sorted into a 96-well plate and analyzed by time-lapse imaging, allowing parallel quantification of expression kinetics and emergent functions. To demonstrate the platform's potential, we first screened computationally re-designed variants of the bacterial cell division protein FtsZ, identifying variants with improved bundling phenotypes and faster kinetics. We then extended our screening procedure towards general protein modulators of FtsZ and identified a combination that anchors filaments to the interface, producing a ring-like phenotype. PUREdrop bridges computational protein engineering and synthetic cell research, elevating the rational engineering of complex biological function to the next level.
|
|
Scooped by
mhryu@live.com
May 9, 4:50 PM
|
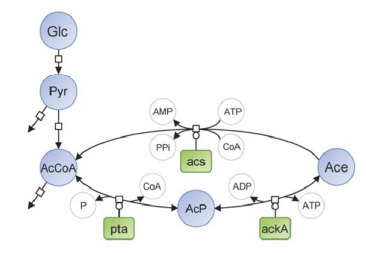
The long-held view that acetate, one of the main fermentation by-products of E. coli, is toxic to microbial growth is currently challenged. Here, we demonstrate that acetate promotes E. coli adaptation to nutrient changes by accelerating growth resumption, with as little as 250 μM acetate being sufficient to shorten the lag phase by several hours. Acetate was found to be consumed via acetyl-CoA synthetase very early after the nutrient change. Transcriptomics, metabolomics and 13C-isotope labeling experiments show that acetate replenishes metabolic pools in the tricarboxylic acid cycle and upper glycolysis. Single-cell analyses reveal that acetate increases the adaptation speed of individual cells switching to the new nutrient. We conclude that the reuse of excreted acetate by E. coli facilitates metabolic adaptation by transiently replenishing central metabolite pools. This work identifies an unexpected role of acetate in the nutritional adaptation of E. coli, providing new insights into the physiological relevance of overflow metabolism.
|
|
Scooped by
mhryu@live.com
May 9, 4:15 PM
|
The eukaryotic genome is non-randomly organized within the nucleus, with positioning linked to function. Still, genome-wide radial maps are missing for the majority of experimental model systems. We adapted Genomic loci Positioning by Sequencing (GPSeq) to Saccharomyces cerevisiae, enabling high-resolution mapping along the nuclear cente–periphery axis. GPSeq confirms known spatial features and shows that peripheral telomeres and centromeres impose long-range constraints extending up to 200 kb, restricting short chromosome arms from the nuclear interior. Telomere repositioning to the nuclear center, either artificially or during quiescence, reorganizes much of the genome through inward movement of sub telomeric regions and compensatory shifts of mid-arm chromatin outward. In quiescence, reduced centromere peripheral localization further alters genome organization. While transcription has a modest impact on radial positioning in all studied conditions, we uncover that in the absence of centromere or telomere constraints, GC–content functionally organizes chromatin in the nucleus.
|
|
Scooped by
mhryu@live.com
May 9, 3:15 PM
|
With the upgrading of global health demands and safety controversies surrounding artificial sweeteners, natural sweeteners have emerged as a research hotspot thanks to their low-calorie content, high safety, and potential physiological activities. Traditional plant extraction methods are limited by bottlenecks such as long growth cycles and geographical dependence, while synthetic biology has provided an innovative pathway for the efficient and sustainable production of natural sweeteners through metabolic engineering and enzyme engineering, by constructing microbial cell factories. This review systematically summarizes the metabolic pathways and microbial synthesis methods of 12 representative natural sweeteners (including sweet proteins, terpenoid glycosides, flavonoids, polyols, and monosaccharides). By analyzing the advantages and disadvantages of different synthetic schemes as well as the core technical bottlenecks in current production processes, and integrating the latest research progress, this review provides theoretical support and technical references for the future industrial optimization and production of natural sweeteners.
|
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
Genetic code expansion introduces new-to-nature chemical moieties into ribosomally synthesized proteins. In practice, the scope of functional groups that can be accessed using this method is often limited by noncanonical amino acid (ncAA) availability. Producing ncAAs directly in cells can circumvent poor ncAA uptake or commercial unavailability, but limited enzymes suitable for this application exist. In vitro evolution campaigns have been remarkably successful in yielding synthetically useful "ncAA synthases." However, these enzymes are optimized for preparative-scale synthesis and their activities often do not translate well to cellular biosynthesis. Thus, expanding strategies to engineer enzymes specifically for ncAA production within cells will benefit further implementation of genetic code expansion. Here, we use phage-assisted noncontinuous and continuous evolution to evolve enzymes for improved synthesis of non-canonical tyrosine derivatives in E. coli. Using simple serial passaging, we uncovered mutations that doubled the production of an expensive ncAA, 3-methoxytyrosine, by tyrosine phenol lyase, and furthermore evolved variants that enable 3-iodotyrosine biosynthesis, a transformation the parent enzyme is unable to catalyze. Additionally, we evolved a recently reported tyrosine synthase for improved production of 3-halogenated tyrosines, identifying variants that exhibit high activity even at low substrate concentrations owing to a ~8-fold reduction in KM. Our results demonstrate that phage assisted evolution can be used to rapidly improve the activity of enzymes for ncAA production in cells.
|
|
Scooped by
mhryu@live.com
Today, 12:47 AM
|
Bioprinting promises on-demand organs, but the reality is costly. Bioprinting promises on-demand organs, but the reality is costly. From expensive bioinks and failed prints to clinical translation hurdles, every step demands significant financial and scientific investment. Recognizing these costs upfront is critical for setting realistic expectations, attracting sustainable funding, and driving meaningful progress in the field.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
Small open reading frames (smORFs), which encode proteins under 100 amino acids, represent an underexplored dimension of the human gut microbiome, despite growing evidence of their essential biological roles. Due to small size and poor annotation, smORFs are typically excluded from metagenomic/metaproteomic analyses. Here, we present a high-resolution multi-omic workflow that integrates smORF prediction into metaproteome searches and enables ultra-deep detection of smORF-encoded proteins (SEPs), without experimental size-based enrichment, utilizing state-of-the-art mass spectrometry instrumentation. Applied to human gut microbiomes, this approach resulted in the largest number of detected SEPs to date, allowing identification of over 25,000 SEPs in the metaproteome, alongside the measurements of the larger proteins. Our multi-omics integrative strategy is critical for advancing human metaproteome research. It also provides a generalizable strategy for comprehensive SEP discovery across diverse microbial ecosystems greatly expanding the previously hidden proteomic landscape. Here, the authors develop a new multi-omics workflow that reveals millions of predicted small open reading frames that translate to thousands of identified small proteins by metaproteomics within the human gut microbiome, expanding understanding of microbial biology and enabling more comprehensive metaproteome studies.
|
|
Scooped by
mhryu@live.com
Today, 12:21 AM
|
Capturing greenhouse gases and converting them into biochemicals is beneficial. Previously, we reversed methanogenesis by cloning methyl-coenzyme M reductase (Mcr) from an unculturable population of anaerobic methanotrophic archaea into the methanogen Methanosarcina acetivorans to capture both methane and carbon dioxide to produce acetate, lactate, and electricity. In the present study, we found that the methane captured (130 ± 30 μmol) by ANME-1 Mcr in M. acetivorans is converted from acetate to ethanol (120 ± 10 μmol). Moreover, ethanol production was tripled (370 ± 20 µmol) by adding iron(III) and humic acids to facilitate electron transfer and yeast extract to stimulate growth. We further introduced heterologous carboxylic acid reductase and alcohol dehydrogenase into the same host with the goal to produce even more ethanol from the intermediate acetate, but we found that this actually leads to a drop in ethanol yield, showing that the methanogenic host converts acetate from Mcr-captured methane by utilizing its native aldehyde ferredoxin oxidoreductase and alcohol dehydrogenase. Hence, we demonstrate that M. acetivorans may be utilized to produce ethanol from methane and carbon dioxide, that this conversion is enhanced by heterologous production of ANME-1 Mcr, and that the host can produce ethanol from acetate. Acetate produced by methanogens from captured methane and carbon dioxide can be converted efficiently to ethanol, demonstrated by lab experiments on the engineered methanogen Methanosarcina acetivorans.
|
|
Scooped by
mhryu@live.com
Today, 12:12 AM
|
Brown algal cell walls are complex matrices composed primarily of alginate, cellulose, and fucoidan. Their depolymerization is important in marine carbon cycling. Although numerous algal polysaccharide-degrading enzymes have been characterized, most studies focus on breaking down single, purified polysaccharides, leaving the degradation mechanisms of native cell walls containing mixed polysaccharides poorly understood. Here we report the integrated modular enzymes involved in brown algal cell wall polysaccharides (BACWPs) degradation. Using the marine flavobacterium Aquimarina sp. 2-A2 as a model, we isolated a bifunctional enzyme, CelAly, which integrates a glycoside hydrolase family 5 cellulase domain and a polysaccharide lyase family 31 alginate lyase domain within a single polypeptide, enabling the degradation of cellulose and alginate in brown algal cell walls. In vivo relevance of CelAly was confirmed by upregulation of its gene during growth on algal biomass. CelAly also contains three distinctive substrate-binding modules (B1, B2, UKD) that support its multimodular functionality; among these, UKD is notable for its dual substrate-binding capability. CelAly’s modular architecture and interdomain flexibility may facilitate coordinated degradation of BACWPs. Bioinformatic analyses and biochemical validation revealed three additional types of such modular enzymes from marine microbes. CelAly and related modular enzymes are strongly associated with marine environments and exhibit conserved modular strategy for substrate recognition and catabolism. Thus, these enzyme architectures represent a previously unrecognized strategy specialized for BACWPs decomposition. This study elucidates the unique structural and functional adaptations of the integrated multimodular enzymes and highlights their ecological prevalence among marine bacteria, providing insights into natural biomass decomposition.
|
|
Scooped by
mhryu@live.com
May 9, 11:52 PM
|
Aspergillus niger is a versatile biotechnological workhorse whose generalist lifestyle enables the utilization of a wide range of sugar compounds, including hexoses, pentoses, and polyols. Yet most predicted A. niger sugar transporters remain uncharacterized. Here, we characterized three glycerol transporters—GlpA, GlpB, and GlpC—from A. niger. We generated single and double glp deletion strains for physiological analysis, among which the ΔglpA strain showed a clear phenotype with significantly reduced glycerol consumption, indicating that GlpA is essential for glycerol uptake. Furthermore, both growth on glycerol and its uptake were abolished in the ΔglpAΔglpB mutant, while the ΔglpAΔglpC mutant showed no growth phenotype on solid medium but lacked glycerol consumption in liquid cultures. This suggests that GlpB plays a supportive role in glycerol uptake, whereas GlpC affects it indirectly. Nevertheless, heterologous expression of the A. niger glp genes in a sugar uptake deficient Saccharomyces cerevisiae strain showed that all three transporters could transport not only glycerol but also D-mannitol, D-sorbitol, and D-xylitol. In addition, GlpC transported the hexose sugars D-fructose, D-glucose, and D-mannose. These results demonstrate the broad specificities of the A. niger Glp transporters, highlighting the need for thorough physiological and functional characterization of filamentous fungal sugar transporters. Our findings advance the understanding of polyol and sugar uptake in A. niger and provide candidate transporters for strain engineering towards biotechnological upcycling of glycerol.
|
|
Scooped by
mhryu@live.com
May 9, 11:39 PM
|
Controlling collective behavior in the microscale is essential for advancing autonomous robotic systems in complex environments. While biohybrid microrobotic swarms offer considerable promise for targeted therapeutic and remediation applications, their programmable assembly and collective behavior remain challenging. Here, we describe an attractive light-triggered approach for enabling reconfigurable swarming of biohybrid microrobots based on the green microalga Chlamydomonas reinhardtii (CR). Such reversible swarming behavior is realized by combining the wavelength-dependent assembly ability of CR and its inherent phototactic properties with light exposures through a series of different mask openings that define the desired swarm geometry. Changes in the projected light enable dynamic modulation of the swarm shape and size, including real-time swarm splitting and merging behaviors. The concept was explored toward artificial intelligence–assisted wound targeting applications through the creation of microrobot swarms customized to exposed wound areas. Such powerful swarming capabilities offer considerable promise for the collective behavioor of biohybrid microrobots toward important practical applications.
|
|
Scooped by
mhryu@live.com
May 9, 11:14 PM
|
Microbes are powerful cell factories for making molecules that are difficult or impossible to produce by other means. Filamentous fungi such as Trichoderma reesei are superior hosts for recombinant protein production, yet secreted proteases often degrade target proteins, reducing yields and limiting process robustness. Preventing proteolysis without relying on expensive commercial inhibitors or compromising strain fitness has been a longstanding challenge in fungal biotechnology, particularly for scaling up production to industrially relevant levels. Here, we report the identification of a native inhibitory protein from T. reesei, TrI9, and show that directing its secretion into the culture medium markedly reduces extracellular protease activity and enables production of the highly protease-sensitive spider silk-like protein CBM-AQ12-CBM, which could be used in high-performance biomaterials. Computational and in vitro analyses provide mechanistic insight, showing that TrI9 functions as a multi-target inhibitor of subtilisin-like proteases (SLPs), including SLP2, a protease that cannot be eliminated by gene deletion due to its crucial role in normal growth and development. This TrI9-based strategy for protease mitigation presents a novel approach for strain improvement, protecting a wide range of protease-labile products beyond silk proteins during large-scale fermentation. Protecting sensitive proteins without added inhibitors offers a cost-effective alternative for scaling up protein production across multiple applications, from protein-based materials manufacturing to pharmaceuticals, as well as enzyme and food applications.
|
|
Scooped by
mhryu@live.com
May 9, 5:11 PM
|
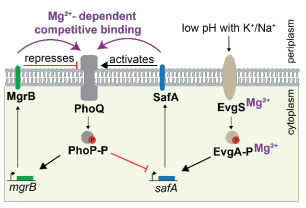
Small proteins are emerging regulators of bacterial virulence via signaling pathways. However, how multiple small proteins coordinate to control a single pathway remains unclear. Here we show that two antagonistic small proteins, SafA and MgrB, forming a magnesium-dependent control module, enhance the signaling sensitivity of the PhoQ/PhoP two-component system in E. coli. Leveraging conditions that enable sequential expression of SafA and MgrB, we dissect and characterize their regulatory dynamics. Our results reveal magnesium as the central control parameter modulating the abundance and affinities of the small proteins for the sensor kinase PhoQ. This coupling together with the competitive binding of the small proteins enables self-adjustment of the signaling network and enhances its sensitivity to environmental magnesium changes. Functionally, loss of either small proteins influences the ability of enteropathogenic E. coli to evade macrophage phagocytosis, linking this regulatory scheme to host interaction. Our findings establish a framework for achieving a high level of input sensitivity through antagonistic regulations in a signaling network.
|
|
Scooped by
mhryu@live.com
May 9, 4:56 PM
|
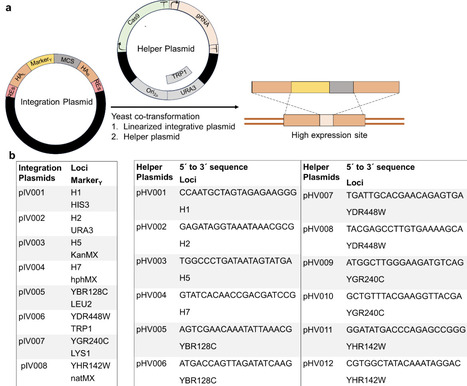
Budding yeast Saccharomyces cerevisiae is a workhorse chassis for producing added food and agricultural compounds. However, building multi-enzymatic pathways for these chemicals often requires iterative genomic integration, underscoring the need for efficient, rapid genome-editing tools that can reliably target transcriptionally active chromosomal regions. In this study, to accelerate strain construction, we established a genome-editing toolkit to rapidly engineer eight loci, highly expressed hot-spots, but nonessential genomic sites suitable for stable pathway assembly. Our approach integrates three key design features: (i) selectable markers to enable rapid screening of edited cells, (ii) extended homology arms that leverage the yeast homology-directed repair machinery for robust genomic integration, and (iii) co-delivery of Cas9 and guide RNAs to promote efficient double-stranded DNA breaks at specific integration sites. The sequence independence of FASTOP relies on the release of integration cassettes from integrative vectors, mediated by restriction digestion at two flanking multiple-cutting sites in the integration module to minimize the risk of introducing sequence errors during PCR amplification of the integration cassettes. Following the introduction of a fluorescent reporter cassette, we observed high integration efficiencies across the target sites. We then integrated the biosynthetic pathway of plant-derived flavonoid naringenin into the hot-spots of the yeast genome using the FASTOP toolkit. Our results demonstrated that upon expressing the five essential genes in simple shake flask culture, naringenin production reached 505.7 mg/L, representing a significant (69-fold) increase over previously reported titers for comparable minimal heterologous pathways in S. cerevisiae. Together, the FATSOP toolkit provides a user-friendly platform for reliably modifying hot-spot loci to rapidly construct multi-enzymatic metabolic pathways in S. cerevisiae, while achieving high production levels for high-value food-relevant metabolites.
|
|
Scooped by
mhryu@live.com
May 9, 4:35 PM
|
Crop nutrition depends on plant-microbe interactions, yet it remains unclear whether conserved genetic pathways impose universal rules on root microbiome assembly across plant hosts. Here, we show that the Common Symbiosis Signalling Pathway (CSSP), a conserved genetic module controlling endosymbiosis with arbuscular mycorrhizal fungi and nitrogen-fixing bacteria, regulates root microbiome assembly in a host-specific manner across contrasting fertilization regimes. Using Lotus japonicus and Hordeum vulgare, we demonstrate that mutations in orthologous CSSP genes remodel root bacterial communities in both species, but with distinct taxonomic outcomes. In Lotus, CSSP disruption reduces rhizobial colonization and promotes niche replacement by commensal taxa, whereas in Hordeum, the same mutations broadly restructure bacterial lineages without converging on Lotus-like responses. Root exudate profiling reveals host-specific metabolic differences, particularly in phenylpropanoid (flavonoids and coumarins) and gibberellin pathways, linking CSSP activity to chemically distinct rhizosphere environments that correlate with divergent microbiome assembly patterns across hosts. Moreover, root bacterial community composition accurately predicts plant nutritional status, highlighting tight coupling between host physiology and microbiome composition. Together, our results show that conserved symbiosis signalling regulates root microbiome assembly, while host-specific metabolic environments determine taxonomic outcomes. This extends CSSP function beyond canonical endosymbioses and positions symbiosis signalling as a general determinant of plant-microbiome interactions with implications for crop nutrition.
|
|
Scooped by
mhryu@live.com
May 9, 3:23 PM
|
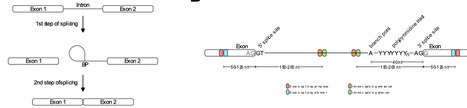
Variants affecting RNA splicing are a major contributor to human disease, yet the consequences of variants outside of the canonical splice motifs are often difficult to determine. Here, we present a protocol for minigene-based evaluation of candidate splice-altering variants. The methodology described includes locus-specific insert design, commercial gene fragment synthesis, and long-read sequencing. The combined approach enables rapid assay development and nucleotide level resolution of the effect on splice isoforms in vitro, providing a scalable framework for functional validation of predicted cryptic splice variants.
|



























compartmentalization