 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:59 AM
|
Gram-negative phytopathogenic bacteria employ a Type III Secretion System (T3SS) to deliver effector proteins (T3Es) into plant cells. While the role of T3Es in suppressing plant immunity is well-established, mounting evidence indicates that these proteins also function as sophisticated manipulators of host physiology and metabolism. This review summarizes recent advances in understanding how T3Es modulate cellular processes beyond immune signaling to establish a conducive niche for colonization. In this review, we examine how effectors facilitate pathogen entry and induce water-soaking by manipulating stomatal dynamics, plant development, and apoplastic hydration. Furthermore, we highlight how bacteria secure nutrients by activating susceptibility genes and reprogramming metabolism, while simultaneously remodeling the host cytoskeleton and vesicular trafficking to dismantle cellular architecture.
|
|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
Nutrients are key drivers of microbial community structure, yet we lack a data-driven quantitative framework linking nutrient environments to community assembly. Here, using controlled microcosm experiments, we systematically probe the effects of nutrient number, concentration, and type on community diversity and structure. To explain these effects, we construct a minimal consumer-resource model incorporating resource competition and cross-feeding. We find that cross-feeding network structure is critical: only shallow, wide networks, where several byproducts are produced from a few supplied nutrients in a few trophic layers, reproduce a linear increase in diversity with the number of supplied nutrients. We also perform new experiments varying nutrient concentration and reveal that diversity slowly decreases with increasing concentration. We explain this finding by incorporating consumption-dependent toxicity into the model, consistent with spent-media measurements. This extended model not only recapitulates virtually all observed patterns but makes an independent prediction: at high nutrient concentrations, communities should be enriched in bistable species pairs, which we confirm experimentally. Our work demonstrates that minimal data-driven consumer-resource frameworks, systematically constrained by experiments, can unify and predict a broad range of nutrient-community relationships.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
Imaging carbon movements in the rhizosphere is fundamentally limited by high soil heterogeneity, low signal levels, and lack of methodology. We present Rhizo-PET, a dedicated positron emission tomography (PET) imaging and analysis framework designed to characterize the 4D spatiotemporal patterns of tracer distribution in intact plant-soil systems. The system achieved a global energy resolution of 11.93 ± 0.02% FWHM at 511 keV and maintained stable performance over 8 h of continuous acquisition, with a coincidence rate variation of only 0.7%. Spatial resolution reached 1.06 mm near the center of the field of view, establishing a high-fidelity region for root-scale analysis. Dynamic datasets were acquired from live Phaseolus vulgaris plants (N = 3) over 180 min following 11CO2 pulse labeling and reconstructed into 3 min temporal frames. Quantitative analysis across 243 independent regions of interest (ROI) revealed that cumulative tracer accumulation decreases monotonically with radial distance from the root axis, while axial transport delays increase systematically in lower root segments (p < 0.001). Hierarchical variability analysis showed that within-plant spatial organization (CVTTP = 0.03) is significantly more stable than inter-plant variation (CVTTP = 0.14), proving that the observed heterogeneity reflects biological spatial organization rather than experimental instability. These results establish Rhizo-PET as a robust, reproducible platform for the non-invasive, time-resolved analysis of carbon dynamics in the rhizosphere under realistic soil conditions.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
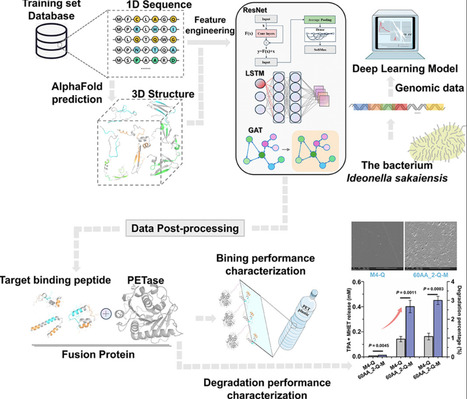
Enzymatic polyethylene terephthalate (PET) degradation holds promise for environmental restoration. However, limited substrate catalytic capacity hinders its application in addressing PET plastic contamination. To enhance enzyme–substrate interaction, effective anchoring strategies are essential. This study presents a novel deep learning approach to identify and engineer high-performance PET-binding peptides from genomic data. Utilizing this approach, we discovered promising PET-binding peptides from the Ideonella sakaiensis genome and fused them with an optimized Ideonella sakaiensis PETase mutant to enhance PET hydrolysis. Remarkably, the Efficient Attention-Based Model for Computational Protein Design-optimized fusion proteins achieved a 2.0- to 24.8-fold increase in PET hydrolysis compared with the enzyme without an anchor. Importantly, we elucidated the mechanism by which the binding peptide domain enhances the catalytic activity of the enzyme against PET substrates, supported by comprehensive molecular dynamics simulations. This work establishes a robust deep learning framework for biocatalyst design and provides potent enzymatic solutions to address global PET plastic pollution.
|
|
Scooped by
mhryu@live.com
Today, 12:37 AM
|
Protein glycosylation in prokaryotes shows extraordinary diversity including species-specific monosaccharides, non-canonical attachment sites, and variable glycan architectures that challenge existing glycoproteomics approaches. Current strategies are largely tailored to eukaryotic systems and depend on predefined glycan databases or prior biochemical knowledge, limiting their application to microbes. Here we present NovoGlyco, a modular glycoproteomics platform for untargeted characterisation of prokaryotic protein glycosylation from shotgun proteomics data. NovoGlyco integrates de novo oxonium ion discovery, sequence tag matching, and mass offset binning to identify novel glycans, their composition, and linking chemistry. An interactive dashboard allows exploration of glycan features and modified proteins. We demonstrate the NovoGlyco platform across published glycoproteomics datasets, spanning human pathogens, Asgard archaea, and environmental enrichment cultures, and identify previously unreported flagella O-glycans in the opportunistic pathogen Campylobacter fetus. In summary, NovoGlyco provides a scalable framework for unbiased exploration of microbial glycoproteomes in both single-organism and metaproteomic contexts.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
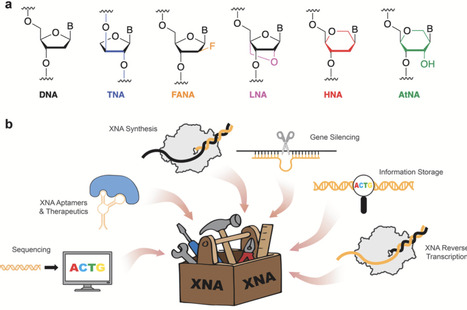
Polymerases are the molecular machines of information transfer, yet their natural catalytic repertoire is largely restricted to DNA, RNA, and a limited set of chemically modified analogs. In response to this constraint, custom polymerases have emerged as powerful tools in the synthetic biology toolbox. Advances in polymerase engineering are unlocking access to RNA polymers generated by primer-extension rather than a promoter driven process as well as synthetic genetic polymers with unnatural backbone structures, collectively termed xeno-nucleic acids (XNAs). In this review, we highlight examples of how custom polymerases are redefining the chemical boundaries of genetic function and driving innovation across the fields of synthetic biology, biotechnology, and molecular medicine.
|
|
Scooped by
mhryu@live.com
April 16, 11:56 PM
|
Microbiomes are now recognised as the second genome of eukaryotes, providing diverse life-support functions for their hosts. The impact of microbiome members on the growth and health of their hosts is determined by chemical cues from the host that modulate microbial physiology, virulence, and the biosynthesis of specialised metabolites. In this review, we provide a cross-kingdom comparison of the role of human and plant molecules in regulating bacterial gene expression. We highlight specific feedback loops and discuss common mechanisms of bidirectional cueing in human- and plant-associated bacteria. Despite the different taxonomies of human- and plant-associated bacteria, we find striking functional similarities in the chemical dialogues at the crossroads of host–bacteria interactions.
|
|
Scooped by
mhryu@live.com
April 16, 11:34 PM
|
Spatial organization plays a critical role in shaping microbial community structure and function, influencing ecological stability, resource utilization, and evolutionary dynamics. Microbial interactions such as competition and cooperation are key drivers of spatial patterning, yet the environmental factors modulating these interactions remain incompletely understood. Here, we investigated how toxic substrates influence the spatial organization of synthetic microbial communities engaged in metabolic cross-feeding. Using a synthetic Pseudomonas stutzeri consortium consisting of the detoxifier and consumer that cooperatively degrade the toxic compound salicylate, we found that increasing the substrate concentration leads to a distinct shift in spatial organization: the detoxifier increasingly dominates the outer periphery of the expanding colony, forming a “detoxifier-first” succession pattern. Mathematical modeling further revealed that this spatial arrangement emerges from substrate toxicity, which selectively favors the detoxifier. Substrate toxicity inhibits consumer proliferation. However, the detoxifier, capable of degrading the substrate, locally reduces toxicity and creates a protective microenvironment that enables nearby consumer cells to survive and grow. In return, the consumer provides essential final products that support the growth and expansion of the detoxifier. This reciprocal interaction establishes a directional dynamic in which the detoxifier, favored by its detoxification capability, colonizes first, paving the way for subsequent consumer proliferation. Our findings demonstrate that substrate toxicity is a crucial environmental factor shaping spatial organization and diversity in microbial communities. This study highlights the importance of considering both metabolic interactions and substrate properties in understanding microbial ecology.
|
|
Scooped by
mhryu@live.com
April 16, 5:03 PM
|
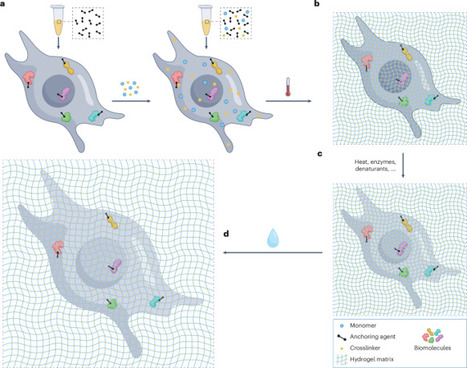
Expansion microscopy (ExM) is a sample transformation technique that enables the resolving of nanoscale biological features on ordinary microscopes. By physically expanding biological samples permeated by a hydrogel matrix, ExM effectively overcomes the diffraction limit of light microscopy, enabling nanoscale effective resolution on conventional imaging systems (such as confocal microscopes) without specialized super-resolution optics. Since its inception, ExM has evolved beyond fluorescence microscopy, enhancing the resolution of techniques such as mass spectrometry imaging, Raman imaging and in situ sequencing, by physically decrowding biomolecules. This Primer aims to provide a comprehensive guide to ExM, focusing on both its most common and latest variants. We outline a modular approach for understanding the scope of ExM protocols, as well as for understanding how to optimize or tune ExM for specific biological questions, covering essential laboratory equipment, sample preparation techniques, imaging techniques and computational approaches. Additionally, we discuss strategies for handling and analysing the vast data generated by ExM, highlight key applications across various biological fields, including cellular and structural biology, neuroscience, pathology and microbiology, and address current limitations. Finally, we explore emerging innovations in expansion chemistry, multimodal imaging and automation that will further broaden the impact of ExM across biological and clinical research. Expansion microscopy enhances conventional imaging by physically enlarging samples within a hydrogel matrix, surpassing diffraction limits. In this Primer, Vo et al. detail typical expansion microscopy protocols, optimization strategies and applications across various biological disciplines, while addressing limitations and future innovations in imaging techniques and data analysis.
|
|
Scooped by
mhryu@live.com
April 16, 4:34 PM
|
Many questions remain about the biochemical mechanisms of quorum sensing (QS), particularly in Gram-positive bacterial pathogens that pose serious threats to human health such as Staphylococcus aureus. Methods to isolate and study the molecular components of these signaling systems in vitro are not straightforward. To date, most mechanistic investigations into QS systems have been accomplished in cells and, to a lesser extent, using chemical inhibitors. Herein, we report the development and characterization of a set of engineered Bacillus subtilis strains that can be used to study the accessory gene regulator (agr) QS circuits of a variety of Gram-positive bacteria. We cloned the S. aureus agr QS machinery into B. subtilis as proof-of-concept to generate “sender” cells capable of autoinducing peptide (AIP) signal biosynthesis, “receiver” cells capable of signal transduction, and “full” cells containing the full agr QS system. We verified that the B. subtilis sender cells could produce the native S. aureus AIP and that the receiver and full cells responded to both the AIP signal and a panel of known chemical agr modulators. The approach was readily transferable to study the agr QS systems of Staphylococcus epidermidis and Listeria monocytogenes, revealing interesting differences that could indicate underlying divergences in native QS mechanisms. We also demonstrate the ability of these B. subtilis strains to function as biosensors to detect the native AIPs of bacteria and produce a targeted antibiotic in response. These engineered systems should find utility for the study of QS in a range of fundamental and applied contexts.
|
|
Scooped by
mhryu@live.com
April 16, 4:00 PM
|
Bacterial persister cells that exhibit a transient state of antibiotic tolerance play a key role in chronic and recurring infections. Despite advances in our understanding of persisters, many aspects of their phenotypic plasticity, particularly their metabolism, are poorly characterized. In this Perspective, we examine the static and dynamic characteristics of persister metabolism that are shaped by genetics, environmental cues, and the intrinsic variability in cellular processes. These factors underlie much of the diversity observed among persister cells and largely explain the inconsistency in expression of classic persister hallmarks such as biphasic killing curves or metabolic dormancy. Further, the literature suggests that persisters are not uniformly metabolically dormant but represent a range of metabolic states. We will focus on the unique rewiring of the metabolic mechanisms in persisters, which depends on both internal and external factors. Bacterial persister cells exhibit a transient state of antibiotic tolerance and are commonly assumed to be metabolically ‘dormant’. In this Perspective, Orman et al. re-examine common assumptions and emphasize that persisters represent a range of metabolic states, driven by internal and external factors, which may explain the inconsistent expression of classic persister hallmarks.
|
|
Scooped by
mhryu@live.com
April 16, 3:51 PM
|
Plants face constant environmental stresses that induce conflicts between growth and defense. The rhizosphere microbiome helps resolve this conflict by enhancing nutrient-uptake efficiency and activating plant immunity simultaneously. In this review, we first outline the mechanisms and limitations of plant-intrinsic growth-defense trade-offs; we then describe the integrated support that rhizosphere microbial communities provide for plant nutrition and defense. Finally, we propose the “microbial-damper” framework and further elucidate the interactions and feedback mechanisms that constitute this system. The microbial damper is a conceptual framework describing the capacity of the rhizosphere microbiome to stabilize a plant’s internal growth-defense resource allocation, which is otherwise perturbed by stresses such as nutrient imbalance and other environmental stresses. This framework highlights how the rhizosphere microbiome can reduce stress-induced plant growth-defense resource-allocation conflicts, thereby providing actionable strategies for designing sustainable agricultural systems.
|
|
Scooped by
mhryu@live.com
April 16, 3:41 PM
|
Codon sequences can influence proteins to misfold during cotranslational folding. Here, we develop an in vivo assay in E. coli to comprehensively study the impact of single synonymous substitutions on protein folding efficiency and apply it to the N-terminal domain of ddlA. By mapping the influence of codons along the sequence, we show that codon identity can substantially influence folding efficiency in a manner depending on structure and topology. We found that a cluster of codons in the N-terminal domain strongly impacts ddlA folding. Further analysis revealed that substitutions to rarer codons generally lead to increased folding efficiency. Consistent with this, an mRNA composed exclusively of rare codons yields higher expression and folding efficiency than one containing only commons codons. Our results highlight the importance of rare codons in cotranslational folding and the relationship between codon sequence and protein structure.
|
|
|
Scooped by
mhryu@live.com
Today, 1:43 AM
|
Cable bacteria are filamentous microbes that couple sulfide oxidation to oxygen reduction over centimeter distances via long-distance electron transport. While their activity creates characteristic biogeochemical gradients that shape sediment ecology, the study of cable bacteria has been constrained by the chemical and physical heterogeneity of the natural sediments they inhabit. To date, laboratory cultivation efforts have relied on these undefined environmental matrices. Here, we established a reproducible enrichment and cultivation platform using an artificial sediment matrix coupled with chemically defined media. This matrix successfully supported the growth of both freshwater and marine cable bacteria and enabled serial propagation over multiple transfers. Microsensor profiling confirmed that the incubations recapitulated hallmark geochemical signatures, including the sulfide, oxygen and pH gradients associated with electrogenic sulfur oxidation. Scanning electron microscopy confirmed the presence of cable bacteria, while 16S rRNA sequencing confirmed enrichment of the cable bacteria together with a stable co-enriched community that included taxa associated with sulfur and iron cycling as well as cellulose decomposition. This defined cultivation system eliminates the variability inherent to natural samples, providing a controlled platform for dissecting the physiology, genetics, and microbial interactions of cable bacteria.
|
|
Scooped by
mhryu@live.com
Today, 1:28 AM
|
Root exudation mediates the delivery of plant primary and secondary metabolites into soil, where they regulate plant-microbe interactions and terrestrial carbon cycling. Conventional exudate analyses quantify total root-released carbon, yet obscure the spatial origin and rhizosphere influence of individual compounds. Here, we develop a rhizobacterial biosensor platform, named Suc-MAPP, to map local exudate profiles along the surface of colonized root tissues. Focusing on sucrose, we engineered sfGFP-based, sucrose-responsive gene circuits in Pseudomonas putida KT2440 for live imaging of exudate concentrations in the micromolar range. These biosensors reveal spatially structured sucrose exudation patterns across eudicots and monocots and implicate photoassimilate source-sink dynamics as a major determinant. We further apply this platform to phenotype exudation modulated by synthetic gene circuitry in Arabidopsis thaliana, identifying genetic design rules for graded sucrose release and quantifying how engineered export sculpts rhizosphere assembly of a defined bacterial community. Together, these results establish programmable rhizobacterial biosensors as tools to spatially resolve plant-environment carbon exchange in situ and provide a framework for extending this approach to diverse exudate targets.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
2,4-Dihydroxybutyric acid (DHB) is a promising C4 platform compound for the synthesis of methionine analogues and biodegradable polymers. However, aerobic DHB production from glucose in E. coli involves transient acetate overflow prior to product synthesis, which could be challenging for process scalability. Therefore, we engineered E. coli K-12 MG1655 for optimized DHB production by replacing the phosphotransferase system mediated glucose uptake with the galactose permease GalP, coupled to ATP-dependent phosphorylation via endogenous glucokinase. In combination with targeted deletions of malate- and fumarate-consuming reactions, we obtained a strain with enhanced flux through the tricarboxylic acid (TCA) cycle and pentose phosphate pathway leading to improved NADPH availability and increased anaplerotic activity, as revealed by 13C metabolic flux analyses. Deletion of the mdh gene encoding for the cytosolic malate dehydrogenase further promoted DHB formation. The resulting strain achieved DHB yields up to 0.20 mol mol−1 (2.43 g L−1), a 4-fold increase compared to the wildtype background (0.05 mol mol−1, 0.60 g L−1), under aerobic conditions while suppressing acetate formation. Together, these results demonstrate that GalP-mediated glucose uptake and engineering of the TCA cycle provide a robust metabolic framework for efficient DHB biosynthesis and establish a foundation for further process and pathway development.
|
|
Scooped by
mhryu@live.com
Today, 12:47 AM
|
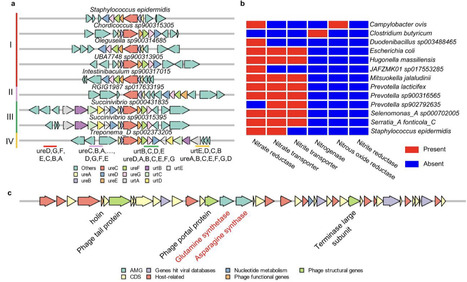
Optimizing nitrogen (N) utilization in ruminant production systems holds both economic and environmental significance. However, traditional paradigms of N metabolism, derived primarily from well-studied model rumen bacteria, do not fully reflect the diverse and complex N metabolism in the rumen ecosystem. To address this gap, we utilized comparative genomics and genome-resolved multi-omics analyses using a curated set of microbial genomes to investigate N assimilation and regulation in rumen microbes. We discovered that well-established mechanisms of ammonia assimilation and regulation, such as the glutamine synthetase (GS)/glutamate synthase (GOGAT) pathways and their regulatory proteins, are absent in many of the predominant rumen microbes, which likely utilize alternative pathways for ammonia assimilation. These findings challenge the applicability of E. coli-based N regulation models to rumen bacteria in response to ammonia availability. We further linked polysaccharide utilization and ammonia assimilation across hundreds of rumen microbial species. Furthermore, we identified specific microbial species involved in ureolysis and denitrification, as well as phages carrying auxiliary metabolic genes involved in N assimilation. Using an animal trial involving 11 pairs of lamb twins in a crossover design, we demonstrated that dietary crude protein (CP) at 10% and 13% had minimal impact on rumen microbiome composition and expression of N assimilation genes. Instead, changes in concentrate levels altered N assimilation, notably increasing expression of amino acid biosynthesis pathways. These findings indicate a nuanced, species-specific microbial response to dietary interventions, highlighting the limitations of traditional N metabolism models applied to rumen microbes and the need for more granular studies of rumen microbial ecosystems.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
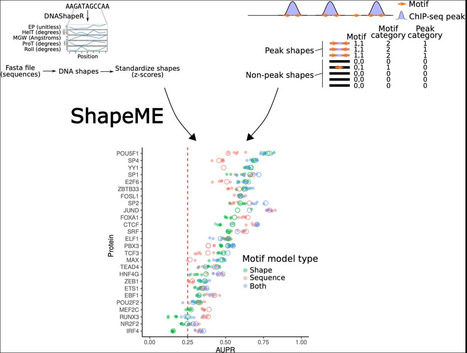
Determining where proteins bind a genome is paramount to understanding gene regulation. In addition to DNA sequence motifs, structural motifs (e.g., a narrow minor groove width) determine binding for some proteins. Algorithms using structural features of DNA to predict protein binding exist, but a structural motif discovery framework which can be applied to a variety of experimental designs is needed. We present a workflow capable of utilizing virtually any type of data representing sequence coverage or enrichment (e.g. ChIP-seq, RNA-seq, SELEX, etc.) to discover structural motifs with explanatory power for a protein’s DNA binding preference. Our approach to motif discovery wraps shape and sequence motif inference into a single tool called ShapeME (github: https://github.com/freddolino-lab/ShapeME.git, web interface: https://seq2fun.dcmb.med.umich.edu/shapeme). Application of ShapeME to ENCODE datasets reveals proteins for which short structural motifs outperform the best PWM for that protein at the JASPAR database, or as identified by the sequence motif elicitation tool STREME. ShapeME is a powerful, versatile framework for inferring structural DNA binding motifs.
|
|
Scooped by
mhryu@live.com
Today, 12:08 AM
|
Identifying molecular binders for protein targets through virtual screening is an active and rapidly expanding field, as the ligands discovered can serve both as molecular probes for mechanistic studies and as initial hits for drug discovery. Since pioneering work in the early 1990s, docking-based virtual screening (DBVS) has become a cornerstone of structure-based drug discovery and has achieved substantial success in identifying novel small-molecule modulators for diverse therapeutic targets. In this review, we first describe the major components of DBVS workflows, including ligand-binding site identification, chemical library preparation, and molecular docking methodologies. We then summarize recent advances aimed at improving DBVS performance, with a focus on template-based approaches, deep learning-based docking and scoring functions, and the emergence of large-scale and ultra-large-scale docking campaigns. Finally, we discuss current challenges and future opportunities for DBVS, outlining key directions for continued methodological innovation and for maximizing the practical impact of virtual screening in early-stage drug discovery.
|
|
Scooped by
mhryu@live.com
April 16, 11:37 PM
|
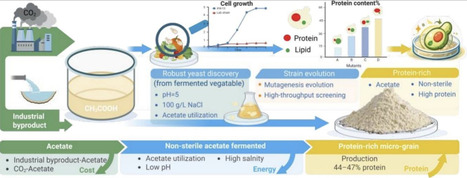
A stable protein supply is essential for both human and planetary health. Low-carbon microbial manufacturing provides an arable land-independent and carbon–neutral route for protein supply, while it is limited by the natural capacity for cellular protein synthesis and high energy consumption for sterile fermentation. To address these challenges, a robust system was established for efficient protein synthesis from low-carbon acetic acid under non-sterile conditions. Yeast strains naturally capable of utilize acetic acid under acid and high-salinity conditions were isolated from traditional fermented vegetables, and their unique ability to enable microbial protein production in non-sterile conditions, mineral medium containing acetic acid as the sole carbon source at pH = 5 or with additional 100 g/L NaCl was validated. To further improve the protein yield, a high-throughput single cell screening method was developed to monitor the cellular macromolecule composition, and acid- and salt-tolerant strains were evolved for higher protein content, achieving an up to 7% improvement. Cultivation of the evolved strain under non-sterile conditions resulted in micro-grains with a protein content of 46 ± 2%. The robustness of the strain and process was further demonstrated through the valorization of low-cost industrial by-product acetic acid into protein. This work explores the potential of natural and artificial evolution and establishes a superior platform for protein-rich micro-grain synthesis through low-cost and low-carbon microbial manufacturing.
|
|
Scooped by
mhryu@live.com
April 16, 11:06 PM
|
Upstream open reading frames (uORFs) are regulatory elements present in the 5′ leaders of mRNA that can significantly impact downstream gene expression in eukaryotes. In crop engineering, editing of uORFs can provide an avenue to upregulate expression of native genes without the need to add persistent transgenic copies. Even with genome-wide methods to identify translated uORFs such as ribosome profiling, their functional characterization depends on validation through reporter gene assays and mutagenesis studies. Current screening methods for plants use luciferases or protoplasts to measure differential gene expression between wild-type and mutated transcript leaders, which requires tissue processing and/or substrate addition. Here, we present a time- and cost-efficient alternative to investigate transcript leaders by co-expression of two fluorescent proteins in Nicotiana benthamiana leaf tissue and test our assay on genes involved in photoprotection, editing of which could provide a pathway to increase CO2 assimilation during sun–shade transitions.
|
|
Scooped by
mhryu@live.com
April 16, 4:55 PM
|
The Enterobacter genus contains 23 species that include common nosocomial pathogens capable of causing a wide variety of infections. We obtained all available Enterobacter genomes and retained 4,805 high-quality genomes after quality control. Genome sequencing analysis of Enterobacter species revealed the presence of type VI secretion systems (T6SS) in these bacteria, but systematic analysis and comparison of these systems among different species are limited. We found that these bacteria code for three distinct types of T6SS, each with a unique set of diverse predicted effectors. Whereas at least 14 effectors are found in each strain, the number of immunity proteins is considerably fewer. By demonstrating a correlation between the abundance of known T6SS-associated proteins and the presence of T6SS, we proposed a comparative genomics model to evaluate the correlation between unknown T6SS-associated ortholog proteins and T6SSs. Among the homologous groups most strongly associated with T6SS, we potentially identified several effectors. It is conceivable that our methodology could be scaled to survey additional bacterial genera for novel T6SS effectors, thereby providing fresh perspectives and directions for subsequent biological experiments.
|
|
Scooped by
mhryu@live.com
April 16, 4:18 PM
|
“Evasion of cell death” is a hallmark of cancer, enabling transformed cells to withstand oncogenic and therapeutic stress. Restoring cancer cell death is an appealing strategy but requires a deep understanding of cell death programs. Over the past two decades, the cell death field has expanded from apoptosis to include necroptosis, pyroptosis, ferroptosis, and other emerging programs, reshaping cancer biology and revealing therapeutic opportunities. While apoptosis remains the primary radiation- and chemotherapy-induced cell death program, non-apoptotic programs can drive inflammatory responses and orchestrate the interplay among tumor, stroma, and immune components, influencing immunotherapy outcomes. Ferroptosis, an iron-dependent, lipid peroxidation-driven cell death modality, lacks a canonical induction signal and arises from perturbations in lipid, iron, and redox metabolism. This review presents a unified framework for understanding the roles of major cell death programs in cancer development, progression, and treatment response, as well as addressing resistance to cancer cell death and immune suppression.
|
|
Scooped by
mhryu@live.com
April 16, 3:58 PM
|
Staphylococcus aureus Cas9 (SaCas9) is smaller than the widely used Streptococcus pyogenes Cas9 (SpCas9) and has been harnessed for gene therapy using an adeno-associated virus vector. However, SaCas9 requires a longer NNGRRT (where N is any nucleotide and R is A or G) protospacer adjacent motif (PAM) for target DNA recognition, thereby restricting the targeting range. Although PAM-relaxed Cas9 variants have been developed, expanded targeting is often accompanied by compromised target specificity. Here, we report the rational engineering of eSaCas9-NNG, a SaCas9 variant that recognizes relaxed NNG PAMs while maintaining high target fidelity, thereby overcoming a fundamental trade-off in Cas9-based genome editing. eSaCas9-NNG efficiently induces indels and base conversions at endogenous sites bearing NNG PAMs in human cells and mice, with editing efficiencies comparable to those of other PAM-relaxed nucleases, including SpRY, SpG, and iGeoCas9, but with reduced off-target activity. We further determine the cryo-electron microscopy structures of eSaCas9-NNG in five distinct functional states, revealing the structural basis for its relaxed PAM recognition, improved target specificity, and nuclease activation. Overall, our findings demonstrate that eSaCas9-NNG could be used as a versatile genome editing tool for in vivo gene therapy, and improve our mechanistic understanding of the diverse CRISPR-Cas9 nucleases. CRISPR-Cas9-based genome editing is powerful but limited by target range, specificity, and delivery constraints. Here, authors engineer a compact SaCas9 that recognizes NNG PAMs for efficient genome and base editing in cells and mice, and reveal its activation mechanism via cryo-EM structural analysis.
|
|
Scooped by
mhryu@live.com
April 16, 3:46 PM
|
The high specificity of phages toward their hosts holds great promise for phage therapy while posing a challenge to computational prediction approaches. To this end, we introduce PhageHost (P&H), an ensemble pipeline for large-scale discovery of phage tail fibers and strain-level host prediction in Klebsiella pneumoniae using protein language models (PLMs). The pipeline begins with TailSeek, a PLM for tail fiber detection from phage and prophage genomes. Building on TailSeek predictions, we developed HostBuster, a deep learning framework that integrates tail fiber features with host-specific information to predict the lytic potential of phage-K. pneumoniae pairs. In silico and experimental validations confirm that P&H achieves high sensitivity and precision in both tail fiber identification and host prediction. Moreover, our framework demonstrates strong generalizability, enabling large-scale mining of tail fibers from prophage data across diverse bacterial taxa. P&H significantly accelerates high-throughput phage screening, offering a scalable tool for clinical phage therapy applications.
|




























m-2st