Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:47 AM
|
Protein–protein interactions (PPIs) are fundamental to cellular function and metabolic regulation. Mapping these complex molecular networks is essential for understanding signaling pathways, yet it remains challenging due to their transient nature. We discuss how next-generation proximity labeling is evolving from bulk methods toward precise, dynamic PPI mapping, providing actionable biological insights.
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
Site-specific insertion of gene-sized DNA fragments remains an unmet need in the field of genome editing. IS110-family serine recombinases have recently been shown to mediate programmable DNA recombination in bacteria by using a bispecific RNA guide (bridge RNA) that simultaneously recognizes target and donor sites. In this work, we have shown that the bridge recombinase ISCro4 is highly active in human cells and provided structural insights into its enhanced activity. Using plasmid- or all-RNA–based delivery, ISCro4 supports programmable multikilobase excisions and inversions and facilitates donor DNA insertion at genomic sites with efficiencies that exceed 6%. Last, we assessed ISCro4 specificity and off-target activity. These results establish a framework for the development of bridge recombinases as next-generation tools for editing modalities that are beyond the capabilities of current technologies.
|
|
Scooped by
mhryu@live.com
Today, 1:04 AM
|
Training physical neural networks directly in matter remains difficult because most platforms do not implement weight storage and weight update within the same physical substrate. Here we show that engineered E. coli can implement a genetically encoded local learning rule acting on a persistent biological memory. In memregulons, analogue weights are stored as plasmid copy-number ratios in a coupled two-plasmid system and are rewritten by activity-dependent growth bias under a global negative learning signal. In single-strain cultures, theory predicts that the change in mean weight is proportional to the activity of the learning channel and to the standing variance of the stored distribution, and flow-cytometry trajectories across eight distinct promoters driving the learning channel support this prediction quantitatively. At the single-cell level, repeated negative learning also reshapes the stored distribution by narrowing it and increasing its skewness as weights approach the lower boundary. In mixed populations and nine-strain co-cultures, one global negative learning signal selectively rewrites only the active memregulons, enabling supervised adaptation in a bacteria-versus-bacteria tic-tac-toe tournament. We then generalise this principle across nine orthogonal chemical inputs and combinatorial promoters, including channels controlled by quorum-sensing molecules, and use it to rationally design a biological XOR gate. Finally, we examine multilayer ANN-like architectures with a human-in-the-loop protocol in which weight updates remain physically implemented and parameterised by experimental measurements, while inter-layer communication is supplied externally. These results establish a route to physical learning in living matter and provide a modular foundation for adaptive multicellular computation, paving the way for autonomous biological hardware capable of distributed environmental sensing and next-generation cellular therapeutics.
|
|
Scooped by
mhryu@live.com
Today, 12:42 AM
|
Protein-function prediction is crucial for elucidating molecular mechanisms driving biological processes and therapeutics development. Despite numerous computational tools demonstrating promising performance, they fall short when predicting rare, uncharacterized functions or indirect activities. Here, we present COSMOS, a context-aware Gene Ontology (GO) subgraph mining system for protein-function prediction. By leveraging inductive subgraph foundation models and an enriched knowledge graph of protein-GO relationships, COSMOS performs zero-shot, few-shot, and low-homology protein-function prediction. Built on 7,923,952 functional semantic relationships, COSMOS demonstrates robust capabilities to (1) generate state-of-the-art predictions for GO classes with sparse or no experimental annotations, (2) provide interpretable functional subgraphs for transparent rationale analysis, and (3) deliver complementary benefits when integrated with existing embedding-based prediction methods. We anticipate that COSMOS will serve as a complementary approach to conventional protein annotation methods and an interpretable tool for predicting protein functions within underexplored GO classes, thereby advancing genomics and therapeutic research.
|
|
Scooped by
mhryu@live.com
Today, 12:05 AM
|
The ability to access, search, and analyse large collections of RNA molecules together with their secondary structure and evolutionary context is essential for comparative and phylogeny-driven studies. Although RNA secondary structure is known to be more conserved than primary sequence, no existing resource systematically associates individual RNA molecules with curated phylogenetic classifications. Here, we introduce PhyloRNA, a curated meta-database that provides large-scale access to RNA secondary structures collected from public resources or derived from experimentally resolved 3D structures. PhyloRNA allows users to search, select, and download extensive sets of RNA molecules in multiple textual formats, each entry being explicitly linked to phylogenetic annotations derived from five curated taxonomy systems. In addition to taxonomic information, each RNA molecule is accompanied by a rich set of descriptors, including pseudoknot order, genus, and three levels of structural abstraction - Core, Core Plus, and Shape - which facilitate comparative analyses across sets of molecules. PhyloRNA is publicly available at https://bdslab.unicam.it/phylorna/ and is regularly updated to incorporate newly available data and revised taxonomic annotations.
|
|
Scooped by
mhryu@live.com
March 19, 11:58 PM
|
RNA is frequently chemically modified, with over 170 types of chemical modifications identified to date in cellular RNAs. These modifications, along with their effector proteins, constitute new layers of gene expression regulation by controlling either the fate of modified RNAs at nearly every stage of their life cycle or local transcription through modulating the nearby chromatin state and transcriptional complexes. This is especially evident in dynamic biological contexts such as cellular state transitions, signaling, immune responses, and stress adaptation. In this review, we discuss recent breakthroughs and promising avenues for future exploration. Particular attention is given to the functional significance of mRNA modifications, the emerging roles of modifications on chromatin-associated regulatory RNAs in chromatin and transcriptional regulation, and mechanistic insights that will guide future scientific interrogation of RNA modifications in gene expression regulation. We also highlight how these fundamental understandings are beginning to catalyze the development of novel therapeutic strategies.
|
|
Scooped by
mhryu@live.com
March 19, 11:33 PM
|
Polyhydroxyalkanoate (PHA) synthases are a group of complex, dimeric enzymes which catalyze polymerization of R-hydroxyacids into PHAs. PHA properties depend on their monomer composition but enzymes found in nature have limited specificities to certain R-hydroxyacids only. In this study, a conditional variational autoencoder was used for the first time to design novel PHA synthases. The model was trained with native protein sequences obtained from Uniprot and was used for the creation of approximately 10 000 new PHA synthase enzymes. Out of these, 16 sequences were selected for in vivo validation. The selection criteria included the presence of conserved residues such as catalytic amino acids and amino acids in the dimer interface and structural features like the number of -helices in the N-terminal part of the enzyme. Two of the 16 novel PHA synthases that had substantial numbers of amino acid substitutions (87 and 98) with respect to the most similar native enzymes were confirmed active and produced poly(hydroxybutyrate) (PHB) when expressed in yeast S. cerevisiae. The results show the power of AI based methods to create active variants of highly complex dimer enzymes.
|
|
Scooped by
mhryu@live.com
March 19, 11:07 PM
|
A major challenge in microbiome research is the inherent complexity and inter-individual variability of the human gut microbiota. To address this, we have developed a detailed protocol for establishing and analyzing a Simplified Human Intestinal Microbiota(SIHUMI)—a defined, in vitro bacterial consortium composed of seven fully sequenced and anaerobically culturable human gut commensals. This model enables highly reproducible and controlled experiments, in which the individual growth of each member can be quantitatively tracked over time (up to 48 h) via species-specific qPCR. The protocol outlines optimized and standardized steps, including consortium setup, time-resolved sample collection, DNA extraction and qPCR analysis. It can be used to evaluate community dynamics in response to interventions such as nutrients, antimicrobials or other xenobiotics. The system is readily adaptable: additional strains can be incorporated, including pathogens (e.g., Clostridioides difficile), to transform it into an infectious disease model. In addition, we describe two optional rapid methods for assessing interspecies interactions and provide an open-source web app for generating interaction network plots. This enables exploration of ecological mechanisms and potential off-target effects. The entire workflow—from setup to data acquisition—can be completed within 1 week. This qPCR-based protocol offers a validated and accessible platform for gut microbiome research, providing a standardized, strain-level and time-resolved alternative to 16S- or fluorescence-based workflows and enabling quantitative, scalable analysis of defined microbial communities. This protocol enables users to establish a defined, in vitro consortium composed of seven fully sequenced and anaerobically culturable human gut bacteria and follow the growth of individual members via strain-specific qPCR to evaluate community dynamics in response to interventions.
|
|
Scooped by
mhryu@live.com
March 19, 10:54 PM
|
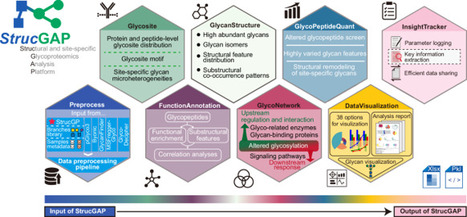
The rapidly developing search engines for glycopeptide identification and accumulated high-resolution glycoproteomic data underscore the need for robust downstream data mining platforms towards subsequently functional and mechanistic studies. Here, we introduce StrucGAP, a Structural Glycoproteomics Analysis Platform for scalable downstream data mining of site-specific N-glycoproteomics. It integrates modules for data quality control, overall glycan structural characterization, altered glycan feature extraction, functional annotation, as well as upstream regulation and downstream networks. Its visualization and insight-tracking functionalities distill interpretation across hundreds of outputs, uniquely enabling to generate chart-based analysis reports and extract key glycosylation insights—capabilities rarely found in existing omics tools. Applying StrucGAP to an uncharacterized aging mouse uterus dataset reveals bidirectional regulation of core-fucosylation, and progressive, coordinated enrichment of glycans featuring sialylation via Neu5Ac, Lewis epitopes, and hybrid glycans along glycosylation dynamics. These changes are functionally linked to adhesion and remodeling, demonstrating StrucGAP’s ability to distill critical glycosylation insights from multi-dimensional information of structural N-glycoproteome datasets. StrucGAP is a structural glycoproteomics analysis platform that distills multi-dimensional information of glycoproteome datasets into structural and functional insights towards mechanistic studies.
|
|
Scooped by
mhryu@live.com
March 19, 10:30 PM
|
Our understanding of how membrane asymmetry governs biological function is limited by the lack of techniques to produce model membranes which can reliably and accurately mimic cellular membrane asymmetry. Not only in terms of asymmetric lipid distribution, but also how that asymmetry can be confined to specific lateral locations across the membrane. Here we present an inverted emulsion method that can be used to produce synthetic cells with symmetric and asymmetric bilayers, as well as phase separation where the intermembrane domains possess distinct bilayer asymmetries. We assess the degree of lipid asymmetry using protein-lipid interaction and quenching assays. Surprisingly, the synthetic cells with asymmetric and phase separated membranes displayed pronounced curvature of the domains and resulted in membrane budding and division. Overall, this work develops biomimetic membranes with lipid compositions akin to natural biomembranes – an essential element in the development of functional synthetic cells. Lipid bilayer asymmetry, phase separation, and encapsulation are long standing issues in the development of synthetic cells. Here the authors develop an inverted emulsion method that can produce synthetic cells with symmetric and asymmetric bilayers, as well as phase separation with domains possessing distinct bilayer asymmetries.
|
|
Scooped by
mhryu@live.com
March 19, 5:21 PM
|
Terminal-based workflows are central to large-scale structural biology, particularly in high-performance computing (HPC) environments and SSH sessions. Yet no existing tool enables real-time, interactive visualization of protein backbone structures directly within a text-only terminal. To address this gap, we present StrucTTY, a fully interactive, terminal-native protein structure viewer. StrucTTY is a single self-contained executable that loads mulitple PDB and mmCIF files, normalizes three-dimensional coordinates, and renders protein structures as ASCII graphics. Users can rotate, translate, and zoom in on structures, adjust visualization modes, inspect chain-level features and view secondary structure assignments. The tool supports simultaneous visualization of up to nine protein structures and can directly display structural alignments using Foldseek's output, enabling rapid comparative analysis in headless environments. The source code is available at https://github.com/steineggerlab/StrucTTY
|
|
Scooped by
mhryu@live.com
March 19, 5:18 PM
|
Zymomonas mobilis is an ethanologenic Alphaproteobacterium with many interesting characteristics for fundamental research and applied microbial engineering. Although genetic engineering has been established for Z. mobilis since the 1980s, a rich set of inducible transcriptional regulators is still unavailable. In this work, seven different chemically inducible promoters have been systematically tested for their functionality in Z. mobilis. In particular, for the first time, NahR-PsalTTC, VanRAM-PvanCC, CinRAM-Pcin and LuxR-PluxB have been characterized in Z. mobilis, alongside the commonly used regulator-promoter pairs TetR-Ptet and LacI-PlacT7A1_O3O4, and the less commonly used XylS-Pm. All promoters investigated in this work are compatible with the Golden Gate modular cloning framework Zymo-Parts. Characterization was carried out with a shuttle vector backbone based on pZMO7, which has so far been rarely used for applications in Z. mobilis but seems to be completely stable without selection and generates high and uniform levels of expression. From the experimental results presented, it can be concluded that VanRAM-PvanCC and CinRAM-Pcin are particularly promising for broad use in the Z. mobilis community.
|
|
Scooped by
mhryu@live.com
March 19, 4:34 PM
|
Bacteria in the human gut influence host physiology and disease risk, but their ecology is strongly shaped by mobile genetic elements (MGEs) such as phages and plasmids. Past interactions between bacteria and MGEs can be inferred from CRISPR-Cas cassettes, which contain short DNA fragments derived from invading elements. To lay the groundwork for research on the impact of such interactions on the human host, we examined bacteria, MGEs, and CRISPR-Cas in the gut microbiome. Using fecal shotgun metagenomes from 1034 Norwegians, we constructed an extended microbiome resource comprising 1.7K prokaryotic mOTUs, 19.5K viral vOTUs and 24.2K plasmid PTUs. We also recovered 74.2K unique CRISPR-Cas cassettes to map past bacteria-MGE interactions and assessed their associations with human diet and lifestyle factors. CRISPR-Cas spacers, and which viruses and plasmids they targeted, varied substantially within bacterial species, but were predominantly directed towards cohort-specific MGEs. Moreover, bacteria were more likely to target MGEs present in the same sample, consistent with local exposure. Bacteria also shared more targets within taxonomic families than across families, where mobilizable plasmids were more frequent among the targets. We did not find evidence that CRISPR-Cas spacers were related to characteristics of the human host, beyond the host-bacteria associations. Together, this research provides a large-scale resource and a structured analysis of bacteria-MGE interactions in the gut microbiome, and their contribution to microbial ecosystem dynamics.
|
|
|
Scooped by
mhryu@live.com
Today, 1:36 AM
|
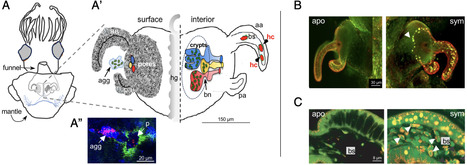
Bacterial outer membrane vesicles (OMVs) and the cargo they carry are increasingly recognized as a means of communication between microbial symbionts and the cells of their host. However, few studies have focused on the biochemical and molecular mechanisms underlying OMV signaling during symbiosis onset and development. We show here that SypC, an OMV protein of the bioluminescent symbiont Vibrio fischeri, is taken up by cells of the squid host Euprymna scolopes where it assumes a new function, i.e., the facilitation of symbiont-induced light-organ morphogenesis. SypC is a Wza-like outer membrane protein found in host-associated Vibrionaceae and is essential for V. fischeri biofilm formation. Colonization or direct treatment with V. fischeri OMVs triggers host development, which was reduced or delayed if the host is instead exposed to a ∆sypC mutant or ∆sypC OMVs. RNA-seq analyses comparing light organs colonized by either the mutant or its parent revealed differential expression of host genes associated with immune responses and tissue morphogenesis. In immunocytochemical imaging, SypC-bearing OMVs were taken up by the host’s macrophage-like cells near the light-organ crypts, revealing the mechanism by which SypC travels through tissue to trigger morphogenesis. Taken together, the data provide evidence that in addition to its role in biofilm formation and colonization, SypC has a second function promoting the induction of symbiotic-tissue development. These findings provide a critical piece of a puzzle whereby a rich array of host and symbiont molecules work in concert to orchestrate normal symbiont colonization and host development within the first hours to days of symbiosis.
|
|
Scooped by
mhryu@live.com
Today, 1:23 AM
|
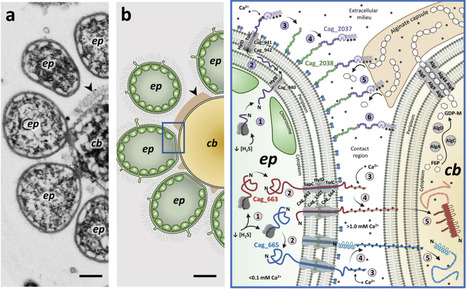
The transfer of virulence factors into eukaryotic cells is a hallmark of bacterial pathogenesis. We report the expression, interspecies transfer, subcellular localization, and potential functions of three unusually large virulence factor-like proteins that underlie a bipartite mutualistic bacterial symbiosis. These proteins are synthesized by green sulfur bacterial epibionts surrounding a central motile chemoheterotroph in the multicellular phototrophic consortium 'Chlorochromatium aggregatum'. While symbiosis-proteins remain intracellular during axenic epibiont growth, they are transferred to the partner bacterium in the association. An RTX-like protein secreted towards the central bacterium is capable of degrading its alginate capsule, thereby promoting direct cell-to-cell contact. Two gigantic hemagglutinin-like proteins are predicted to fold when binding extracellular Ca2+ to form Type 6-like auto injection needles, explaining their observed transfer into the central bacterium. These functionalities extend far beyond the known pathogenic interactions of bacteria with eukaryotes and provide new perspectives on the evolution of bacterial virulence factors.
|
|
Scooped by
mhryu@live.com
Today, 12:46 AM
|
Rice paddies naturally host methane-oxidizing bacteria known as methanotrophs, due to the production of methane in flooded soils. Enhancing the activity of native methanotrophs could improve the sustainability of rice cultivation, but knowledge of how this could impact other members of the rice microbiome remains incomplete. To gain insight into which members of the rice microbiome might benefit from increased methanotrophic activity, we passaged 51 aerobic microbial enrichment cultures from rice rhizosphere and tissue samples in a chemically-defined medium with methane as the primary carbon source and electron donor. We profiled the cultures over time by 16S rRNA gene amplicon sequencing and sequenced the genomes of 44 isolates to gain functional insights. Taxa whose relative abundance increased during community growth on methane represented more than a dozen families, many of which are not known to utilize one-carbon substrates. Several of the enriched genera have not previously been linked to methane cycling in rice fields, and genomic analysis of the sequenced isolates revealed considerable variation in predicted carbon source utilization and nitrogen cycling capabilities. Together, these findings broaden the understanding of how aerobic methanotrophs may impact microbiome assembly and nutrient cycling in rice paddies.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
Plastic biodegradation in natural environments is increasingly recognized as a multi-organism process, yet the mechanisms enabling coordinated depolymerization and metabolism of polyethylene terephthalate (PET) remain poorly understood. Previously, we demonstrated that a full consortium containing three Pseudomonas and two Bacillus strains isolated from hydrocarbon-rich coastal soils of Galveston Bay, Texas, can synergistically depolymerize PET plastic and utilize it as a sole carbon source, a capacity not observed in individual isolates. In this report, using integrated comparative genomics, proteomics, and chemical analyses, we show that PET degradation in this system reflects exaptation of hydrocarbon metabolism reinforced by metabolic division of labor. Within this naturally occurring consortium, Bacillus strains persist under environmental stress, establish biofilms, and perform essential secondary hydrolysis, while Pseudomonas strains catabolize aromatic monomers and buffer oxidative stress. Genes supporting these functions are enriched within the accessory genomes of the consortium strains, indicating consortium-enriched horizontal gene transfer (HGT). In addition to the canonical two-step hydrolytic pathway well documented in PET biodegradation, we identify a secondary methylation- and redox-associated process, mechanisms where the full consortium acts on the oligomer mono(2-hydroxyethyl) terephthalate (MHET), yielding nearly complete conversion to terephthalic acid (TPA) and methylated MHET (MMHET). Together, these findings demonstrate how cooperation and competition within consortia facilitate targeted gene exchange, enabling emergent plastic biodegradation in natural microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:02 AM
|
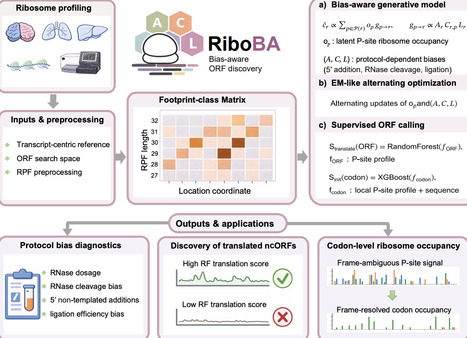
By mapping ribosome-protected fragments (RPFs) genome-wide, ribosome profiling (Ribo-seq) has uncovered extensive translation beyond conventional coding sequences, revealing non-canonical ORFs (ncORFs) with emerging roles in diverse biological processes. However, protocol-induced biases introduced during library construction can substantially distort RPF signals. Most existing ORF callers are not designed to explicitly account for such artifacts, limiting robust ncORF identification. Here, we present RiboBA, a bias-aware probabilistic framework to address this challenge. RiboBA consists of two main components: a generative module that recovers protocol-induced biases and codon-level ribosome occupancy, and a supervised module that identifies translated ORFs and initiation sites using the resulting bias-adjusted profiles. Evaluated through simulations and on a range of Ribo-seq datasets-particularly supported by cell-type-specific immunopeptidomics-RiboBA robustly recovers protocol-induced parameters and achieves superior accuracy and sensitivity in ncORF identification. Notably, RiboBA performs particularly well on RNase I libraries with attenuated three-nucleotide periodicity, as well as on MNase and nuclease P1 libraries, while maintaining competitive runtimes. In a Drosophila case study, RiboBA identifies conserved ncORFs with coding potential, including recurrent upstream translation of ThrRS and Mettl2 that suggests a potential threonine-specific translational control axis.
|
|
Scooped by
mhryu@live.com
March 19, 11:44 PM
|
Attaching and effacing pathogens, including enterohemorrhagic E. coli (EHEC), colonize their preferred intestinal niche by sensing diverse host-, diet-, and microbiota-derived signals and coordinating the expression of virulence factors. D-serine, a host metabolite abundant in urine but scarce in the intestine, restricts EHEC colonization by transcriptionally repressing the type 3 secretion system (T3SS) while activating the SOS stress response. However, the mechanism underlying virulence regulation by D-serine remains unestablished. Here, we show that multiple amino acids, including L-serine converge on this pathway, repressing the T3SS without inducing the SOS response. Transcriptomic analyses showed a common response to D- and L-serine dominated by repression of nitrogen stress response genes. Mutational analysis identified the response regulators NtrC and Nac as essential mediators of T3SS repression by both serine enantiomers. Disruption of L-serine deaminase enzymes crucially revealed that T3SS repression depends on cytoplasmic ammonia/ammonium release rather than sensing of intact serine. While EHEC lacks canonical D-serine catabolic capacity, through metabolomics we provide evidence of oxidative deamination activity, capable of producing this regulatory signal. Together, these findings establish a mechanistic link between amino acid catabolism, nitrogen stress signaling, and virulence regulation in EHEC, highlighting how metabolic flux fine-tunes pathogen adaptation to intestinal niches.
|
|
Scooped by
mhryu@live.com
March 19, 11:25 PM
|
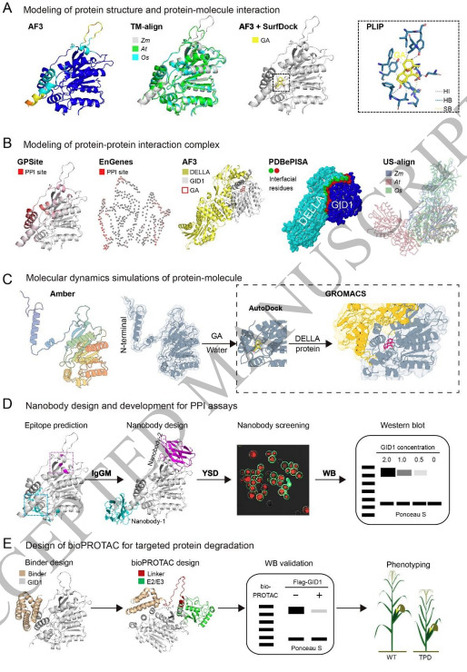
Artificial intelligence (AI) is poised to reshape the research paradigm of the life sciences by rapidly advancing the adoption of protein language models and their derivative tools. These technologies are increasingly being applied to protein structure prediction, function analysis, and protein design throughout the life sciences, and have only recently begun to gain attention within the plant science community. Moreover, while the era of AI-driven bio-breeding is on the horizon, it remains largely in the proof-of-concept stage. Therefore, there is a pressing need not only to outline the fundamental principles, models, and tools in this rapidly evolving field, but also to explore their potential applications in plant research and crop breeding. This review begins by introducing general principles and widely used models for protein understanding and generation, supported by illustrative case studies that highlight how these tools are advancing fundamental plant research. For instance, the analyses of two maize (Zea mays) genes demonstrate how a structure-aware interpretation of the relationships between mutations and protein function enables more precise hypothesis generation and facilitates experimental validation. Subsequently, the review presents generic AI-enabled protein engineering strategies and pipelines, including rational, semi-rational, refactoring, and de novo design, tailored to diverse protein engineering objectives. These approaches aim to create artificial variants and synthetic proteins with improved or novel functions to foster innovation in crop breeding. Finally, the significant challenges of applying protein design in plants are discussed, particularly in light of the limited availability of experimentally resolved protein structures and the inherent complexity of plant biological systems.
|
|
Scooped by
mhryu@live.com
March 19, 11:00 PM
|
Protein structures provide a wealth of information regarding biological functions and underlying mechanisms. The growing availability of high-quality structure predictions and extended molecular simulations has further expanded the potential to leverage these data in a myriad of different ways. Yet, an abundance of data can obscure important information, making it difficult to focus on biologically relevant features. Residue interaction networks (RINs) address this challenge by condensing structural data into subsets of well-defined noncovalent molecular interactions. In this Protocol, we explore how the RIN generator (RING) software can be used to gain biological insights by constructing detailed RINs for proteins and protein–ligand complexes. We provide a step-by-step guide to performing both single- and multi-state protein analyses using the RING web server and a stand-alone software package. In addition, we include a dedicated procedure for sequential multi-file analysis, which can be performed exclusively through the command-line interface. All potential inputs and outputs are explained in detail, along with strategies for downstream data processing. Designed for researchers in biology and related fields with minimal or no programming experience, the entire workflow can be completed in <45 min. Residue interaction networks (RINs) describe noncovalent molecular interactions within and between proteins. This Protocol uses RING software to generate these networks from protein structures toward understanding protein structure and function.
|
|
Scooped by
mhryu@live.com
March 19, 10:35 PM
|
Natural transformation drives the spread of antibiotic resistance among bacteria. The DNA receptor ComEA is essential for transporting external transforming DNA into the periplasm by an unknown mechanism. Here, single-molecule optical tweezers and electron microscopy approaches show that Geobacillus stearothermophilus ComEA forms dynamic oligomers on DNA that can switch between two conformations depending on local concentration. When ComEA sparsely decorates DNA, it forms bridging oligomers that condense the DNA to generate sub-pN pulling forces. When ComEA more fully decorates DNA, it forms non-bridging oligomers that decondense DNA and cannot generate force. Mutating ComEA to favor either bridging or non-bridging conformations causes transformation deficiency in Bacillus subtilis, meaning condensation and decondensation each play mechanistic roles. Our results show that ComEA reversibly condenses DNA during natural transformation, first producing force to pull DNA into the periplasm and then abating force production to promote transport into the cytoplasm. The molecular mechanisms underlying natural transformation remain poorly understood. Here, the authors use optical tweezers to show how the periplasmic DNA receptor ComEA drives the inward pulling of DNA by switching between oligomerization states.
|
|
Scooped by
mhryu@live.com
March 19, 10:25 PM
|
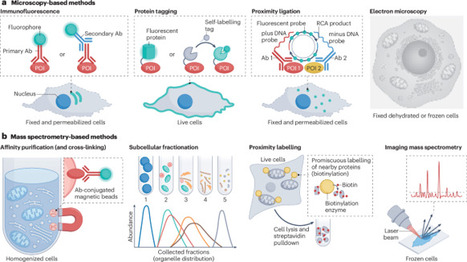
How proteins localize to specific compartments, function in coordination with other biomolecules and, ultimately, contribute to diverse cellular activities are crucial questions in cell biology. Complicating the answers to these questions are multilocalizing and multifunctional proteins, whose impact on the cell depends on both spatial and temporal contexts. Therefore, contextualizing protein functions based on their subcellular localization is necessary to fully understand cell behaviors. Recent advances in instrumentation and protein labelling techniques are rapidly increasing the availability of tools, technologies and applications that measure and control protein localization and compartment-specific function. In this Review, we first discuss microscopy, mass spectrometry-based correlation profiling and proximity labelling methods that assign localizations to proteins, ranging from cellular compartments to protein–protein interactions. We next examine the available tools for manipulating protein localization and measuring the effects of these manipulations, including localization tags and bifunctional molecules. For each technology, we assess the strengths and weaknesses that ultimately determine their usefulness. We conclude with an outlook on future technological advances in the field of spatial subcellular proteomics and their potential implications for cell biology and clinical applications. Subcellular localization is important for protein function. This Review discusses advances in spatial subcellular proteomics, assesses the strengths and weaknesses of technologies that measure and control protein localization, and considers their potential uses in cell biology and medicine.
|
|
Scooped by
mhryu@live.com
March 19, 5:21 PM
|
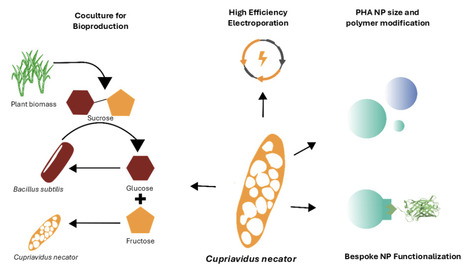
Microbes have the potential to manufacture plastics from sustainable feedstocks while enabling novel material properties and functions that are not easily accessible through conventional chemical synthesis. Realising this potential requires a comprehensive genetic and process engineering framework that spans chassis and bioprocess optimisation, polymer property control, and downstream functionalisation. Here we develop such a platform in Cupriavidus necator, with a focus on high-value polyhydroxyalkanoate (PHA) nanoparticles. To this end we first optimize the transformation protocol for the organism. Next, we create a library of PhaC synthase variants from C. necator, Aeromonas caviae and Brevundimonas sp. in a ΔphaC background, demonstrating that they allow customisation of the material properties of produced PHA particles. Our results combine data from Flow cytometry, Transmission Electron Microscopy (TEM), Fourier Transform InfraRed Spectroscopy (FTIR), and Differential Scanning Calorimetry (DSC) to show that it is possible to generate materials ranging from highly crystalline PHAs to softer P(3HB-co-3HHx) copolymers and that an A. caviae PhaC variant can double the yield of large PHA granules. To improve bioprocess sustainability, we coupled C. necator with B. subtilis in sucrose-fed co-cultures, using tetracycline tolerance differences and inoculation ratios to enhance PHA production from inexpensive, sugar-rich feedstocks. Finally, we add function to the produced PHA nanoparticles by using the molecular protein-fusion technology SpyTag-SpyCatcher, showing it is possible to efficiently capture SpyCatcher-GFP on PHA granules as a proof of concept for PHA′s use as a customisable bio-based nanoparticle. Together, our work offers an innovation to produce bio-PHA nanoparticles in a customisable way, with potential applications in sustainable biomanufacturing, biosensing, drug delivery and future bioremediation technologies.
|
|
Scooped by
mhryu@live.com
March 19, 5:15 PM
|
Many experiments rely on expensive or scarce liquids, such as costly reagents, or biological samples available only in limited quantities. Droplet microarrays are an especially promising approach to conserving these materials because they support highly parallelized reactions in small volumes. However, existing droplet microarray loading methods based on discontinuous dewetting suffer from loading inconsistencies and large dead volumes. In this work, we present the Small Volume Loader (SVL) for the Surface Patterned Omniphobic Tiles (SPOTs) platform that enables precise deposition on droplet microarrays while minimizing reagent waste. By establishing a physical model of the loading process, we identified that deposition volume is governed by the sum of hydrostatic and Laplace pressures at the reservoir outlet. To optimize performance, we engineered a pressure-compensating flared reservoir geometry that maintains constant total pressure regardless of the remaining liquid level. This design ensures that the deposited volume is independent of reservoir volume and reduces dead volume to 5 µL. We demonstrated the platform's utility through high-throughput elicitor screening for natural antimicrobial production from Streptomyces venezuelae. The resulting assays used 100-fold less material than conventional methods, allowing us to conduct over 32,000 assays with modest quantities of starting material. This enabled us to identify specific stressors that optimize the production of the antibiotics chloramphenicol and jadomycin B. Together, we demonstrated improved loading performance for droplet microarray platforms, allowing precise, accessible, and high-throughput assays using only minimal volumes of scarce materials.
|