 Your new post is loading...

|
Scooped by
mhryu@live.com
March 14, 11:32 PM
|
The acid activity of enzymes, characterized by the minimum pH at which enzymes remain active (pHmin), is crucial for industrial applications in acidic environments. However, the rational design of acid-active enzymes remains challenging due to limited understanding of sequence-structure-activity relationships under acidic conditions. Here, we propose ACENet, a graph neural network that predicts enzyme pHmin by integrating surface features of protein structures with evolutionary representations derived from the large-scale protein language model ESM-2. ACENet achieved a Pearson correlation coefficient of 0.85 on the test dataset, significantly outperforming other deep learning baseline models and maintains stable pHmin predictions under various conditions. Even on a subset of the dataset with less than 20% homology, the PCC remains above 0.5, with an RMSE (Root mean square error) less than 1.4. ACENet also present excellent performance in the annotation of pHmin for homologous proteins and the predictive screening of minimal active pH in protein mutants. Remarkably, ACENet could identify the catalytic region as key determinants of acid activity through residue-level interpretability analysis. Overall, ACENet accelerates the development of highly efficient biocatalysts for diverse applications where acidic conditions predominate.
|
|
Scooped by
mhryu@live.com
March 14, 10:37 PM
|
Infertility affects ~1 in 6 people of reproductive age and remains difficult to treat because causes are heterogeneous and diagnostics are incomplete. Recent evidence reframes the female reproductive tract as a low-biomass but biologically active microbial ecosystem. Dysbiosis, typically loss of protective Lactobacillus species (notably L. crispatus) with overgrowth of anaerobic pathobionts, is associated with implantation failure and recurrent pregnancy loss. Framing conditions such as chronic endometritis and reproducible low-Lactobacillus endometrial profiles as dysbiosis-related disorders clarifies opportunities for prevention, companion diagnostics and microbiome-directed therapies. This narrative review contrasts receptive (Lactobacillus-dominant) versus dysbiotic states and summarizes mechanisms linking microbiota to fertility: microbial metabolites (lactic acid, short-chain fatty acids) support epithelial barrier function and immune tolerance, whereas dysbiosis provokes inflammation that impairs implantation. Although observational data consistently associate Lactobacillus dominance with better outcomes, evidence quality is low-to-moderate due to retrospective designs, methodological heterogeneity, and a lack of adequately powered, diagnostic-stratified randomised trials. The review highlights precision microbial therapeutics under development, single-strain next-generation probiotics, synthetic consortia, engineered live biotherapeutics, postbiotics, targeted phage/endolysins and vaginal microbiota transplantation, and proposes a diagnostic-driven roadmap that matches microbiome endotypes and clinical contexts (e.g., preconception vs. immediate embryo transfer) to specific interventions. Regulatory and safety issues for reproductive biologics are also considered. The reproductive microbiome is a promising translational frontier but currently offers a consistent signal rather than definitive proof of benefit. To translate promise into practice requires standardised low-biomass sampling/reporting, mechanistic validation in human-relevant models and diagnostic-stratified randomised trials with staged endpoints, alongside strategies to address engraftment, formulation and regulatory pathways.
|
|
Scooped by
mhryu@live.com
March 14, 10:24 PM
|
The CRISPR–Cas9 system provides adaptive immunity against invading genetic elements through a dual-RNA-guided DNA cleavage mechanism. This system relies on the precise assembly of a ribonucleoprotein (RNP) complex composed of the Cas9 endonuclease, a CRISPR-derived RNA (crRNA), and a trans-activating CRISPR RNA (tracrRNA). Around 100 anti-CRISPR proteins that inhibit CRISPR–Cas systems have been identified, and the mechanisms by which they act are increasingly being elucidated. However, the inhibitory mechanisms of many Acrs, including AcrIIA7, remain poorly understood. Here, we present the structure of AcrIIA7 and uncover a previously unrecognized mechanism by which it inhibits Cas9 function. Structural and biochemical analyses reveal that AcrIIA7 specifically binds to tracrRNA, preventing its association with crRNA and thereby blocking formation of the active Cas9 RNP complex. This tracrRNA hijacking mechanism represents a unique strategy for CRISPR inhibition, in which an anti-CRISPR protein targets an RNA scaffold essential for Cas9 activation rather than interacting directly with the Cas9 protein. Our findings provide the first structural insight into tracrRNA-targeted anti-CRISPR activity and highlight RNA–RNA interaction interfaces as vulnerable nodes in CRISPR–Cas immunity. CRISPR–Cas9 immunity relies on forming a Cas9–crRNA–tracrRNA ribonucleoprotein complex. Here, the authors show that AcrIIA7 blocks this process by binding tracrRNA, preventing its pairing with crRNA.
|
|
Scooped by
mhryu@live.com
March 14, 5:15 PM
|
Jasmonic and salicylic acids are key hormones involved in plant responses to pests and pathogens. Existing fluorescence-based approaches to imaging plant defence hormones are constrained by the need for external illumination and by autofluorescence of plant tissues, while luminescence-based ones require exogenous substrates. Here, we use jasmonate- and salicylate-responsive promoters to engineer autoluminescent plants that report hormone signalling activity with up to a 53-fold contrast. Using consumer-grade cameras, we image reporter Arabidopsis thaliana and Nicotiana benthamiana plants throughout normal development and in response to pest and pathogen attacks, visualising local and systemic responses. Because the luminescence is self-sustained, these reporters enable non-invasive, substrate-free imaging of defence signalling over extended time courses without specialized equipment. Salicylic acid and jasmonic acid are two major hormones regulating plant response against pests and pathogens. Here, the authors configured the autoluminescence pathway to be conditional on activities of these hormones, achieving direct visualisation of plant defense responses.
|
|
Scooped by
mhryu@live.com
March 14, 5:09 PM
|
Across phyla and hosts, morphological plasticity serves as a common strategy for overcoming host defenses and optimizing growth. Whether toggling between yeast and hyphae to invade tissues, as seen in C. albicans and O. novo-ulmi, or shedding the cell wall for concealment like E. muscae, structural transitions are crucial in shaping the pathogenesis of these fungi. While efforts to block morphogenesis in C. albicans are underway, the broader importance of fungal “shapeshifting” remains underappreciated. Morphological plasticity is a recurring strategy that underlies the virulence of diverse fungal pathogens. Indeed, while the three examples we highlighted exploit yeast-hyphal transitions, there are other morphological transitions that fungi leverage for pathogenesis. Cryptococcus neoformans, for example, forms Titan cells—massive, multinucleate fungal cells that bud to produce normal sized daughters—while Coccidioides species form large, endospore-filled spherules. Understanding how pathogenic fungi regulate diverse morphological transitions could have far-reaching implications for developing effective antifungal treatments across phyla.
|
|
Scooped by
mhryu@live.com
March 14, 5:03 PM
|
Archaeal antiviral defense systems remain poorly characterized despite recent advances in understanding prokaryotic immunity. Here, we analyze 7747 archaeal genomes, the largest and most diverse dataset to date, revealing a striking disparity in defense system prevalence and diversity compared to Bacteria. Nearly one-third of archaeal genomes have no detected systems beyond CRISPR-Cas and restriction-modification (in contrast to only 2.2% bacterial genomes), and only 50–55% contain CRISPR-Cas systems, far below previous estimates. Many known defense systems appear restricted to Bacteria, while several single-gene putative candidate systems (PDCs) recently identified through a guilt-by-embedding approach are enriched in Archaea. Phylogenetic analyses suggest that PDC-S70 and PDC-M05 likely originated in Archaea, representing rare archaeal contributions to the prokaryotic immune repertoire. Consistent with earlier studies, our findings support the existence of deep evolutionary links between archaeal and eukaryotic systems for argonautes and viperins. These analyses highlight both the underexplored nature and the evolutionary significance of archaeal immunity, calling for expanded efforts to uncover archaeal-specific systems and improve our understanding of immune evolution across domains of life.
|
|
Scooped by
mhryu@live.com
March 14, 4:54 PM
|
The large size of widely used CRISPR-Cas enzymes limits their delivery for therapeutic applications. Cas12j nucleases offer a hypercompact alternative but show modest editing efficiency. To overcome this limitation, we identified eight novel Cas12j orthologs from viral metagenomes, which in their native form exhibit low editing activity in mammalian cells. We therefore engineered T5 exonuclease-Cas12j fusions, resulting in substantially enhanced genome-editing activity across multiple mammalian cell types, reaching levels comparable to established compact CRISPR-Cas editors. Intriguingly, robust cellular editing occurred in the presence of a previously unrecognized trinucleotide sequence context within the target DNA. Furthermore, we developed Cas12j-based adenine base editors by coupling catalytically inactive Cas12j orthologs with adenine deaminase, enabling efficient A-to-G base conversion in mammalian cells. This study expands the CRISPR toolbox by establishing engineering principles that convert compact Cas12j nucleases into efficient and modular genome-editing platforms well suited for delivery-constrained therapeutic applications.
|
|
Scooped by
mhryu@live.com
March 14, 4:47 PM
|
Foods derived from cultured cocoa, coffee, or other plant cells are nearing commercialization. Given the novelty of the production process and product characteristics, it is advisable to address food safety early during product development. International harmonization of a case-by-case approach for pre-market safety assessments should be pursued.
|
|
Scooped by
mhryu@live.com
March 14, 4:42 PM
|
Biodegradable adhesives, unlike their traditional counterparts, are engineered to bond to biological tissues while naturally degraded over time, thereby eliminating the need for removal procedures and reducing the risk of chronic inflammation. These unique features are particularly suitable for temporary biomedical applications such as wound closure, internal sealing, or integration with electronics for active/passive functions. The adhesive performance arises from the strategic combination of biodegradable polymers and adhesion mechanisms that dynamically interact with tissue surfaces. This review introduces recent advancements in biodegradable adhesives through a mechanism-based framework, focusing on five key adhesion strategies: physical interlocking, hydrogen bonding, catechol chemistry, amine-carboxyl coupling, and covalent bonding via diazirine or isocyanate linkages. For each strategy, representative material systems, functional properties, and biomedical implementations that enable strong, conformal adhesion under wet and physiological environments are highlighted, and with a discussion of current challenges and future directions toward intelligent, multifunctional bioadhesives for clinical uses are concluded.
|
|
Scooped by
mhryu@live.com
March 14, 4:13 PM
|
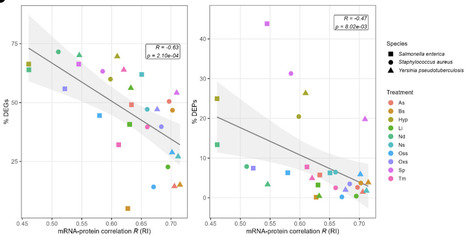
Diverse bacterial pathogens have evolved complex regulatory mechanisms to adapt to various environmental stresses during infection. The uncertainty in mRNA-protein levels in response to environmental stressors complicates our understanding of bacterial physiology and their adaptation to stressful environments. To examine this issue, we have integrated transcriptomics and proteomics data on three human bacterial pathogens Salmonella enterica Typhimurium[CE4.1][KA4.2], Yersinia pseudotuberculosis, and Staphylococcus aureus under ten infection-relevant stress conditions. We observed positive correlations between mRNA and protein levels, which were decreased under different stress conditions. Essential genes exhibited higher expression levels with lower variation across the conditions and stronger mRNA-protein correlations compared to non-essential genes, highlighting their critical role in bacterial adaptability and survival. Moreover, we identified a substantial number of genes with stress-induced non-correlating mRNA-protein levels, particularly under conditions triggering strong stress responses. Particularly this level was dramatically lowered for osmotic stress specific genes affected by impaired translational activity under osmotic stress. Our findings highlight the prevalence of non-correlating mRNA-protein levels and the potential role of post-translational modifications in modulating protein levels in response to environmental stressors during infection. This study provides a comprehensive framework for integrating transcriptomics and proteomics data and identifies potential gene products that might significantly impact the ability of diverse bacterial pathogens to adapt to hostile infection environments.
|
|
Scooped by
mhryu@live.com
March 14, 3:57 PM
|
Computational protein design is often constrained by slow, complex, inaccessible, and highly sophiscated and expert-dependent workflows that hinder its transferrability and generalization power for broader applications. We present ProteinMCP, an agentic AI framework designed to accelerate and democratize protein engineering. ProteinMCP automates end-to-end scientific tasks, delivering dramatic gains in efficiency; for instance, a comprehensive protein fitness modeling workflow was completed in just 11 minutes. This performance is achieved by an AI agent that intelligently orchestrates a unified ecosystem of 38 specialized tools, made accessible through a Model-Context-Protocol (MCP). A cornerstone of the framework is an automated pipeline that converts existing software into MCP-compliant servers, ensuring the platform is both powerful and perpetually extensible. We further demonstrate its capabilities through the successful autonomous design and selection of high-affinity de novo binders and therapeutic nanobodies. By removing technical barriers, ProteinMCP has the potential to shorten the design-build-test cycle and make advanced computational protein design accessible to the broader scientific community.
|
|
Scooped by
mhryu@live.com
March 14, 3:29 PM
|
Genetic code expansion (GCE) enables the site-specific incorporation of noncanonical amino acids (ncAAs) into proteins but is constrained by reliance on exogenously supplied chiral ncAAs. Achieving intracellular ncAA biosynthesis would enable more scalable and cost-effective GCE. Here, we report the continuous hypermutation and evolution of amino acid synthases that produce high levels of ncAAs inside yeast, thus supporting GCE from simple ncAA precursors. We encoded an engineered 'tyrosine synthase' (TmTyrS) on an error-prone orthogonal DNA replication system (OrthoRep) and selected variants based on ncAA biosynthesis from readily available phenol analogs and intracellular L-serine. Our selection employed orthogonal ncAA-specific aminoacyl-tRNA synthetases (aaRSs) as biosensors whereby target ncAA production leads to aminoacylation of an amber suppressor tRNA and the translation of a selectable reporter containing an amber stop codon. Our evolution successfully yielded TmTyrS variants that efficiently produced 3-iodo-, 3-bromo-, 3-chloro-, and 3-methyl-L-tyrosine, enabling amber codon-specified ncAA-dependent translation, in some cases at levels comparable to sense codon-specified natural amino acid translation. This work reduces barriers for expressing proteins containing substituted tyrosines. Moreover, because aaRSs can themselves be evolved (including with OrthoRep) for a flexible range of ncAA specificities, these results establish an end-to-end framework for evolving ncAA biosynthetic enzymes in vivo.
|
|
Scooped by
mhryu@live.com
March 14, 2:24 PM
|
Microorganisms form communities, and their interactions shape the function and stability of these communities. Understanding these interactions can aid in revealing ecosystem dynamics, enhancing community function, and informing the design of synthetic consortia for industrial applications. Deciphering microbial interactions is challenging due to the difficulty of culturing natural microorganisms and the exponential increase in experiments with expanding consortium size. One approach to improving culturing throughput is the use of microcompartments such as agarose microbeads. Microbead-based techniques enable the generation of large numbers of picolitre-sized compartments, facilitating high-throughput, parallel studies of microbial sub-communities. However, the existing microbead-based techniques for deciphering microbial interactions are dependent on single-culture isolates of consortium members and/or labelling of consortium members with fluorescent markers via genetic engineering. We developed a microbead-based, label-free method that eliminates the requirement of single-cell isolates to predict microbial interactions. Our method involves an isolation-independent manner of microbead inoculation with different sub-communities and microbead sorting to separate sub-communities based on growth. Using a probabilistic model, we predict interactions based on cell concentrations and relative abundances in the inoculum and after microbead sorting. We successfully predicted pairwise interactions in two three-member consortia. Additionally, we computationally showcased the validity of our approach for predicting pairwise interactions in larger consortia.
|
|
|
Scooped by
mhryu@live.com
March 14, 10:39 PM
|
With an increasing global cancer burden, the regulatory function of the human microbiome and its metabolites in tumor epigenetics has garnered significant interest. Microbial metabolites are not merely passive byproducts but serve as signaling molecules and epigenetic modulators, contributing to tumor progression through multiple overlapping pathways. Short-chain fatty acids (SCFAs) such as butyrate directly inhibit histone deacetylases to reactivate tumor suppressor genes, while secondary bile acids (BAs) induce gene silencing via DNA methylation remodeling by altering the FXR/TGR5 signaling pathway. Folate and vitamin B12 serve as substrates for DNA and histone methylation through one-carbon metabolism. A complex bidirectional feedback loop exists between microbial metabolism and tumor epigenetics: reprogramming driven by hypoxia or oncogenes alters metabolite flux, generating molecules such as lactate and succinate that not only remodel chromatin and the tumor microenvironment (TME) but also selectively promote the growth of metabolically adapted microbial species, thereby reinforcing epigenetic dysregulation. Despite growing mechanistic insights, establishing causality and correlating spatiotemporal dynamics and dose responses within the highly heterogeneous TME remain major challenges. Data integration across multi-omics remains limited by methodological and computational constraints. Resolving these issues will be critical for understanding the microbe–metabolite–epigenetic axis and advancing personalized precision oncology.
|
|
Scooped by
mhryu@live.com
March 14, 10:28 PM
|
Top-down trophic interactions are major drivers of microbiome dynamics, yet their outcomes are difficult to predict and their consequences for pathogen control remain unclear. We combine synthetic bacterial communities of varying complexity with field studies and microcosm assays to test whether microbivorous nematodes reorganize microbiomes to suppress soilborne disease. Field studies show stronger nematode-microbe associations around healthy plants, and microcosm assays confirm that nematode presence produces stable suppression, whereas microbe-only communities collapse under pathogen invasion. Nematode predation depletes non-preferred bacterial taxa and enriches metabolically versatile taxa within Proteobacteria, increasing community-level antagonistic potential and promoting complementary resource-use interactions linked to pathogen inhibition, yielding suppression beyond individual or pairwise effects. A minimal four-component feedback loop linking a nematode predator, plant pathogens, and two plant-associated bacteria with complementary functions accounts for the emergent outcome. Together, these results reveal an animal-mediated pathway of microbiome assembly that enhances resistance to pathogen invasion and provide a trophically informed framework for designing stable, disease-suppressive microbiomes in agriculture. Top-down trophic interactions are critical drivers of microbiome dynamics but are difficult to predict. Here, the authors demonstrate that nematode-driven shifts in microbiomes enhance pathogen suppression and promote plant health through a feedback loop involving nematodes and bacteria.
|
|
Scooped by
mhryu@live.com
March 14, 5:19 PM
|
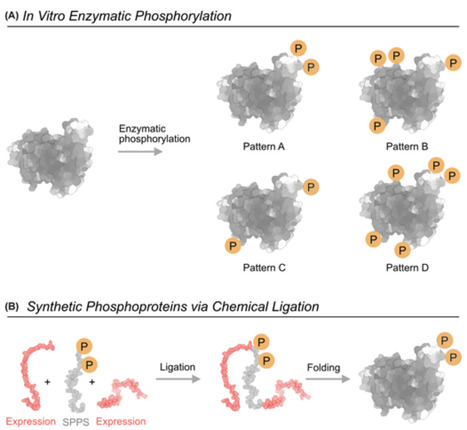
Protein phosphorylation is one of the most common and versatile regulatory mechanisms in cells. Most human proteins are phosphorylated at multiple sites, giving rise to large numbers of possible phosphorylation patterns. Each phosphorylation pattern can lead to a different functional or pathological outcome. Yet, linking defined phosphorylation patterns to specific biological functions remains a major experimental challenge. In this review we describe the main strategies to study phosphorylation patterns at the protein and domain levels and highlight how they complement each other. We first discuss cellular approaches, including phosphomimetics, kinase-based assays, and genetic code expansion, which allow working in a native environment but have their significant drawbacks. We then describe in vitro methods, such as enzymatic phosphorylation and semi-synthetic phosphoproteins generated by ligation, which afford mechanistic insights but result in low yields and are difficult to scale for producing libraries. We focus on synthetic phosphopeptide libraries as tools that offer precise control over the number and position of phosphosites and are uniquely suited for systematic mapping of phosphorylation patterns. This comes at a price of not working at the protein level, but rather at the domain level. Peptide libraries are often used for preliminary identification of key phosphorylations, later studied in detail at the protein level. We conclude that ideally more than one method should be used and that these methods should not be viewed as competing but rather as complementary. A combined use of several of these approaches provides a practical toolbox for dissecting how phosphorylation patterns regulate protein behavior.
|
|
Scooped by
mhryu@live.com
March 14, 5:12 PM
|
Optimization of the design-build-test-learn cycle remains a bottleneck for developing and efficiently manufacturing the next generation of bioproducts. To address this challenge, research has traditionally focused on either data-driven or mechanistic modelling, but the emerging consensus in the field highlights that their strategic integration, often termed hybrid modelling, offers significant advantages, particularly for complex systems where data is sparse. Here, we introduce a hybrid framework where these two methodological lineages are combined to frame development parameters within the metabolism of a production system, therefore achieving mechanism-informed predictions for follow-up experiments. We present three scenarios that exemplify how this framework can be leveraged to guide and accelerate the development of novel bioprocesses, even with small datasets available. We validate our framework by applying it to two heterologous peptide production scenarios in E. coli, explored through commonly relevant experimental factors such as inducer concentration and production strain. Using our framework, we identify key metabolic pathways and reactions that contribute to productivity and whose activity is modified by individual experimental factors like temperature and plasmid used. Furthermore, we show that the biological patterns extracted from this hybrid approach can complement the experimental design, informing predictive models of process performance. Our approach is general and can be tailored to a large array of processes, and thus holds potential for boosting both proof-of-concept and industrial projects, contributing to more efficient and sustainable biomanufacturing.
|
|
Scooped by
mhryu@live.com
March 14, 5:07 PM
|
Quantifying bacteria’s growth rates is essential for understanding their ecological roles and for building predictive models in environmental and clinical settings. Peak-to-trough ratios (PTRs) derived from shotgun metagenomes offer a culture-independent proxy for in situ growth rates of bacterial species, yet their reliable computation remains challenging. We introduce Pilea (https://github.com/xinehc/pilea), an alignment-free, sketching-based method that incorporates statistical models for robust PTR estimation. Pilea achieves speed improvements over existing methods while also enhancing accuracy, as demonstrated on both simulated and real datasets. By scaling efficiently to comprehensive reference collections such as the Genome Taxonomy Database (GTDB), Pilea enables large-scale analyses of bacterial growth dynamics across biomes, unlocking new insights for ecological research.
|
|
Scooped by
mhryu@live.com
March 14, 4:59 PM
|
Horizontal gene transfer (HGT) generates genetic variation in populations across all domains of life; however, most studies focus on individual transfers and functional information derived therefrom. This is useful but does not consider DNA transfer more broadly, that is, nongene transfers, donor–recipient dynamics, or trends and background levels that may help infer ecological information. Here, we review the mechanistic underpinnings of DNA transfer, literature from diverse fields that addresses HGT on a community basis and the associated methodological challenges, and propose a framework for conceptualizing the process of DNA transfer, highlighting DNA mobility as a feature of community ecology and DNA itself as a public good. These ideas coalesce to support DNA transfer as a fundamental ecological phenomenon that remains largely unmeasured.
|
|
Scooped by
mhryu@live.com
March 14, 4:50 PM
|
The stability of synthetic gene circuits is limited by cellular growth and division. Zhang et al. demonstrated how engineered condensates of transcription factors can stabilize gene expression and enhance bio-based production. This work establishes the spatial organization of transcription factors through phase separation as a powerful strategy for robust gene expression.
|
|
Scooped by
mhryu@live.com
March 14, 4:45 PM
|
Robots play an ever-expanding role in society by performing a broad range of tasks. However, there are growing concerns about their environmental sustainability, as many conventional robotic systems rely on materials that are neither renewable nor degradable. Consequently, significant efforts are being made to develop eco-friendly robots built from sustainable and biodegradable materials. In this context, plants represent a promising direction, as the biomaterials composing plants are biodegradable, and their inherent multifunctionality as living organisms, including sensing, actuation, energy harvesting, and self-healing, makes them strong candidates for realizing biodegradable robotic systems. Moreover, they are abundant and renewable resources. Recent studies have demonstrated plant-based robotic systems that harness some of these features, helping to establish plant robotics as an emerging research field. Among the many functions plants offer, actuation is pivotal, as it enables physical robotic motion, such as locomotion and grasping, which substantially broadens the potential applications of plant robots. Focusing on plant movement, this article reviews key plant species and their behaviors through the perspective of actuation characteristics. It also examines the current landscape of plant-based robotic systems and outlines future research directions in this rapidly growing field.
|
|
Scooped by
mhryu@live.com
March 14, 4:21 PM
|
Physiologically relevant biosensors are in increasingly high demand, yet existing ones are severely limited in the number and type of biomarkers that are detected. The lack of biorecognition elements for most medically relevant biomarkers restricts the development of next generation single and continuous use monitors. Over billions of years, microbes have evolved a vast array of proteins to sense and metabolize small molecules, including those pertinent to human health. Of particular interest to us is the identification and subsequent integration of new microbial redox enzymes into electronic biosensors building off the established electrochemical technology of the continuous glucose monitor. Here we deploy genomic screening to identify analyte specific redox enzymes for biosensor development. As a proof of concept, we report the first electrochemical enzyme-based nicotine biosensor from a novel microbial enzyme, and use a variant with improved catalytic performance to enhance sensor performance. The biosensor detects nicotine over 0.4-100 μM, a range relevant to nicotine concentrations present in active smoker sweat, saliva, gastric juice, and urine. This microbial mining approach for discovering redox enzymes expands the sensing parts toolbox available over conventional antibodies and aptamers.
|
|
Scooped by
mhryu@live.com
March 14, 4:04 PM
|
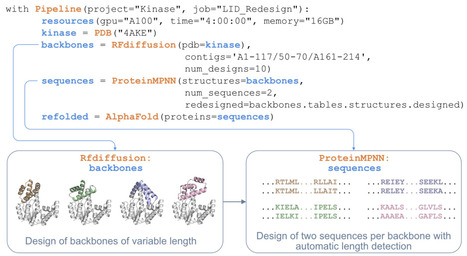
Deep learning methods for protein structure generation, sequence design, and structure and property prediction have created unprecedented opportunities for protein engineering and drug discovery. However, using these tools often requires navigating incompatible software environments, diverse input/output formats, and high-performance computing infrastructure, any of which may hinder adoption by primarily experimental chemical biology laboratories. Here we present BioPipelines, an open-source Python framework that allows researchers to define multi-step computational design workflows in a few lines of code. Additionally, its robust yet modular architecture provides a straightforward way to expand the toolkit with different functionalities, particularly by leveraging coding agents, with little effort. The framework currently integrates over 30 tools encompassing structure generation, sequence design, structure prediction, compound screening, and analysis. The same workflow code can be prototyped interactively in a Jupyter notebook and then submitted for production-scale runs without modification. We demonstrate applications in inverse folding, gene synthesis, de novo protein design, compound library screening, iterative binding site optimization, and fusion-protein linker optimization. We hope this framework will empower researchers, allowing them to focus on the scientific question rather than computational logistics. https://github.com/locbp-uzh/biopipelines
|
|
Scooped by
mhryu@live.com
March 14, 3:54 PM
|
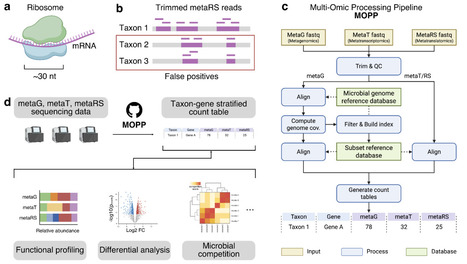
Metaribosome profiling (metaRibo-Seq) enables genome-wide measurement of translation across complex microbial communities by sequencing ribosome-protected mRNA fragments, but the short length of these footprints creates substantial nonspecific mapping against large reference genome collections, leading to spurious taxonomic and functional assignments. Here we present MOPP (Multi-Omics Processing Pipeline), a modular reference-based workflow that denoises meta-Ribo-Seq data by leveraging matched metagenomic coverage breadth to identify genomes likely to be truly present in a sample before aligning metatranslatomic and optional metatranscriptomic reads. MOPP generates taxon-by-gene count tables across genomic, transcriptional and translational layers, enabling integrated downstream analyses of microbial function. We evaluated MOPP using a defined 79-member synthetic human gut community profiled by metagenomics and metaRibo-Seq. Coverage breadth filtering markedly improved detection accuracy relative to a standard baseline workflow, with performance remaining robust across a broad intermediate threshold range and peaking at 92-95% coverage breadth. At a 92% threshold, MOPP reduced the number of distinct detected operational genomic units by 99.4% while retaining 87.8% of aligned metaRibo-Seq reads on average, and increased the F1 score from 0.02 to 0.61. Residual false positives were predominantly attributable to genomes with extremely high nucleotide similarity to true community members, whereas false negatives were enriched among low-abundance taxa, indicating that remaining errors are driven primarily by biological similarity and detection limits rather than widespread nonspecific mapping. Together, these results establish MOPP as a high-throughput workflow for robust processing of metaRibo-Seq in the context of matched metagenomics and position it as a scalable framework for integrated taxonomic and functional analysis of microbial communities across genomic, transcriptional and translational layers.
|
|
Scooped by
mhryu@live.com
March 14, 3:10 PM
|
The PacBio Revio simplifies genome assembly by generating very long reads with very few errors at an affordable price point. Comparative ease of assembly is democratizing access, leading to a larger niche for assembly workflows. HiFi-Helper is a user-friendly snakemake workflow designed to facilitate genome assembly from HiFi data alone. This tool produces a visual summary that provides intuitive feedback on the quality of the resulting assembly and guides assembly parameter optimization. The case studies presented here confirm that HiFi-only assemblies produced by HiFi-helper can meet or exceed the quality of genomes assembled in prior decades with a much lower investment of time and resources.
|






























zengler k