 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:06 AM
|
Many bacterial defense (immune) systems prevent the entry of foreign DNA by directly recognizing and targeting nucleic acids, effectively blocking all mechanisms of horizontal gene transfer. However, systems defending specifically against conjugation, a major route for gene dissemination, have heretofore not been reported. We have discovered a novel defense factor, which we name AbjA (Abortive conjugation protein A), that specifically limits successful plasmid conjugation into a recipient bacterium. AbjA interacts directly with and targets the ATPase component TrbE of the Type IV secretion system (T4SS) to induce cell death; this contrasts with most other defense systems that act at the nucleic acid level. AbjA therefore represents the first member of a new class of bacterial defense factors that trigger what we term "abortive conjugation". Previously, recipient bacteria were viewed largely as defenseless against the mechanism of conjugation; our discovery and characterization of AbjA demonstrates that recipient bacteria can block conjugation to limit the transfer (and thus spread) of plasmids. Discovery of this class of defense systems thus has implications for bacterial defense, plasmid evolution, and possible strategic alternatives to rationally target plasmid spread, particularly with respect to virulence and antibiotic resistance.
|
|
Scooped by
mhryu@live.com
March 11, 11:55 PM
|
Isoprenyl acetate, a volatile ester derived from isoprenol, is a key biosynthetic intermediate for the advanced aviation fuel candidate, 1,4-dimethylcyclooctane. Here, we engineered Pseudomonas putida KT2440 for the production of isoprenyl acetate from mixed sugar substrates. We first generated isoprenyl acetate by introducing a heterologous alcohol acetyltransferase (ATF1) and deleting three promiscuous native esterases to reduce product degradation. Then, we engineered efficient glucose and xylose co-utilization by integrating a heterologous xylose isomerase pathway and deleting global regulators crc and hexRto alleviate catabolite repression. Additionally, intracellular acetyl-CoA flux was reinforced through the expression of auxiliary carbon-conserving routes, including non-oxidative glycolysis and acetate assimilation. Culture conditions were systematically optimized by adjusting medium composition, induction, and overlay solvent to maximize product yields and titers. These cumulative efforts achieved isoprenyl acetate titers of 1.5 g/L in shake flasks and 1.9 g/L in fed-batch bioreactor cultures from mixed sugars, corresponding to a yield of 0.067 g/g of total sugar consumed. Our work demonstrates the potential of P. putida as a robust microbial chassis for scalable biosynthesis of ester-based biofuels from lignocellulosic feedstocks.
|
|
Scooped by
mhryu@live.com
March 11, 11:38 PM
|
Dermatophilaceae polyphosphate-accumulating organisms (PAOs), formerly classified as Tetrasphaera PAOs, play pivotal roles in enhanced biological phosphorus removal (EBPR). However, their phylogenetic diversity, ecological preferences, and metabolic traits remain poorly characterized, and a robust marker gene for their classification is lacking. Here, we performed an extensive phylogenomic and metabolic analysis of Dermatophilaceae PAOs utilizing 46 newly recovered metagenome-assembled genomes from a laboratory-scale EBPR reactor treating high-strength wastewater and full-scale wastewater treatment plants. These analyses revealed a previously uncharacterized PAO genus, named here as Candidatus Dermatophostum, which shows specific preference for high-phosphorus environments. Its representative species, Ca. Dermatophostum ammonifactor, was enriched in the EBPR reactor and its PAO phenotype was confirmed by polyphosphate staining and fluorescence in situ hybridization. Integrative meta-omics combining genomic, transcriptomic, and protein structure analyses revealed its specialized metabolic capabilities for phosphate metabolism, glycogen synthesis, and dissimilatory nitrate reduction to ammonium. Moreover, Ca. Dermatophostum was found to be widely distributed across wastewater treatment plants worldwide, underscoring both its diverse metabolic capabilities and potential engineering implications for mitigating nitrous oxide (N2O) emissions for EBPR system. Finally, we propose a ppk1-based classification framework that resolves Dermatophilaceae PAOs into six distinct clades, consistent with whole-genome phylogeny, and demonstrates that ppk1 can serve as a reliable marker gene for tracking these populations. Together, these findings expand the ecological and functional understanding of Dermatophilaceae PAOs and highlight their promise for advancing sustainable wastewater treatment and resource recovery.
|
|
Scooped by
mhryu@live.com
March 11, 11:26 PM
|
The primary mechanism for transcription termination in bacteria is intrinsic terminators. These terminators influence transcript stability and play key roles in gene regulation. Existing computational methods for genome-wide terminator identification have been designed and evaluated based on a small number of experimentally evinced terminators often from only one or two organisms. We present TerminatorNet, a system for identifying intrinsic transcription terminators throughout bacteria. TerminatorNet uses a neural network model trained on a large set of experimentally characterized transcription terminators from a variety of bacterial genomes. TerminatorNet identifies 98% of terminators and has a false positive rate of 3%, substantially better than existing approaches. TerminatorNet commonly identifies terminators at the ends of operons. We applied TerminatorNet to thousands of genomes across the taxonomic spectrum of prokaryotes, creating a repository of tens of millions of terminators. We observe heavy use of intrinsic termination in some groups, such as Bacillota, and rare use in other groups such as archaea. We also observe a wealth of instances of DNA uptake signal sequences, important components of transformation specificity for some competent bacteria, in terminators identified in Neisseriaceae and Pasteurellaceae.
|
|
Scooped by
mhryu@live.com
March 11, 11:17 PM
|
Rapid gene activation requires transcription factors (TFs) to locate their target motifs within vast genomes. In bacteria, TF target search is accelerated by combining 3D diffusion with 1D sliding, called facilitated diffusion, yet whether eukaryotic TFs rely on similar strategies has remained unresolved due to the lack of direct measurements in living cells. Here, we directly visualize eukaryotic TF target search in living cells by labeling a single TF per nucleus and visualizing its binding to its endogenous locus. Using the budding yeast TF Gal4, we find that efficient target localization occurs near the diffusion limit and does not require facilitated diffusion. Instead, rapid association requires cooperative self-interactions mediated by an intrinsically-disordered central region (IDR), independent of the activation domain. Replacing the Gal4 IDR with human self-interacting IDRs (EWS or FUS) restores efficient search, demonstrating that self-interactions are a general and portable feature for search. A second structured dimerization domain further cooperatives with the IDR to stabilize binding at neighboring motifs, revealing two mechanistically separable forms of cooperativity that together govern TF function. These findings highlight that facilitated diffusion is not strictly required and establish cooperative IDR-driven self-interactions as a key mechanism to enable rapid target recognition in eukaryotic cells.
|
|
Scooped by
mhryu@live.com
March 11, 5:05 PM
|
To address the low efficiency of genetic manipulation and poor hyphal morphology control in Aspergillus oryzae, this study developed a synthetic biology toolkit and identified a key genetic target for morphological engineering. The toolkit features an RNP-mediated rapid knockout system, serine integrase-based gene integration, and a pipeline for screening high-activity neutral genomic sites. Systematic deletion of seven cell wall integrity-related genes revealed that disruption of the chitin synthase gene chsY most effectively enhanced protein secretion. The ΔchsY mutant exhibited a 34.8 % increase in hyphal diameter and a 30.6 % reduction in culture viscosity, coupled with upregulated secretory pathways and an activated unfolded protein response (UPR). Applying this discovery, we engineered a strain expressing a heterologous lipase (TLL), achieving a 52 % increase in extracellular activity in flasks. This benefit scaled to bioreactors, with a 42 % higher enzyme titer and ∼50 % lower viscosity. Our work provides both a genetic toolkit and a scalable engineering strategy (chsY deletion) to enhance A. oryzae as a cell factory for industrial enzyme production.
|
|
Scooped by
mhryu@live.com
March 11, 3:19 PM
|
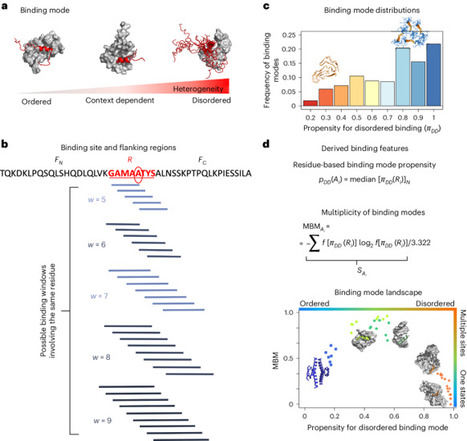
Proteins exhibit complex phase behavior as they convert between the native state, the liquid condensate (or droplet) state and the solid condensate (or amyloid) state. To facilitate the study of these processes, we describe the FuzDrop method of predicting the condensation propensity of proteins to undergo liquid–liquid phase separation and to subsequently form amyloid aggregates. The method is based on the principle that liquid condensations reflect a balance between enthalpic and entropic contributions; FuzPred is an algorithm that provides sequence-based estimates for these contributions in stoichiometric complexes ( https://fuzpred.bio.unipd.it/predictor ). FuzDrop extends this algorithm to protein condensates, and enables prediction of the propensity for amyloid formation within liquid condensates, known as the condensation pathway to protein aggregation ( https://fuzdrop.bio.unipd.it/predictor ). This prediction is based on the principle that the sequence regions that promote aggregation within liquid condensates have a multiplicity of binding modes, because they have a strong propensity for both entropic-driven interactions to stabilize the droplet state and enthalpic-driven interactions to stabilize the amyloid state. The time required for FuzDrop predictions on the web server scales linearly with protein length and is typically ~30 s for a protein of 500 residues. By enabling predictions of protein phase behavior, FuzDrop may facilitate experimental studies directed at the development of therapies for protein condensation diseases. FuzDrop predicts the condensation propensity of proteins on the basis of their amino acid sequences. This protocol describes the underlying theory and how to use the results to understand liquid-liquid phase separation and amyloid aggregate formation.
|
|
Scooped by
mhryu@live.com
March 11, 3:08 PM
|
Bacteria harness diverse defence systems that protect against phage predation, many of which are encoded on horizontally transmitted mobile genetic elements. In turn, phages evolve counter-defences, driving a dynamic arms race that remains underexplored in human disease contexts. For the diarrhoeal pathogen Vibrio cholerae, a higher burden of its lytic phage ICP1 in patient stool correlates with reduced disease severity. However, direct molecular evidence of lytic phages driving selection of epidemic V. cholerae has not been demonstrated. Here, through clinical surveillance in cholera-endemic Bangladesh, we capture the acquisition of a parasitic antiphage mobile genetic element, PLE11, that initiated a selective sweep coinciding with the largest cholera outbreak in recent records. PLE11 showed potent anti-phage activity against cocirculating ICP1, explaining its rapid and dominating emergence. We identify PLE11-encoded Rta as the defence responsible and provide evidence that Rta restricts phage tail assembly. Using experimental evolution, we predict phage counteradaptations against PLE11 and document the eventual emergence and selection of clinical ICP1 that achieve a convergent evolutionary outcome. Finally, we discover how PLEs balance their dependence on ICP1 tail proteins for horizontal transmission with the restriction of phage tail assembly by Rta: PLEs construct chimeric tails composed of both mobile genetic element-encoded and phage-encoded proteins to ensure their transmission. Collectively, our findings reveal the molecular basis of the natural selection of a globally important pathogen and its virus in a clinically relevant context. The acquisition of a parasitic anti-phage mobile genetic element, PLE11, showing potent anti-phage activity against cocirculating ICP1, and the subsequent evolution of ICP1 to escape this defense, are captured, revealing the molecular basis of the natural selection of a globally notable pathogen and its virus.
|
|
Scooped by
mhryu@live.com
March 11, 2:45 PM
|
Microbial serine proteases are valuable for industrial applications due to broad substrate specificity and stability. However, heterologous overexpression in microbial hosts is often limited by cytotoxicity and poor secretion. This study developed an integrated strategy combining protein engineering and signal peptide optimization to enhance extracellular production of PrtA—a key acid-stable alkaline serine protease—in Komagataella phaffii. Directed evolution generated the Q245K variant, showing 1.35-fold higher extracellular expression than wild-type PrtA. A machine learning model, MPEPE (Mutation Predictor for Enhanced Protein Expression), was used to identify critical residues involved in protein secretion; saturation mutagenesis at the top-predicted site generated the I342D mutant with 1.48-fold improved productivity. The double mutant PrtA-Q245K/I342D achieved synergistic enhancement (1.84-fold higher secretion) without altering enzymatic properties. Evaluation of nine signal peptides revealed that serum albumin, α-factor (without pro-region), and PrtA’s native signal peptides each doubled the combinatorial mutant’s secretion, yielding 4.98-fold higher expression than the wild-type. In contrast, α-factor pro-region inclusion drastically reduced yields. In a 15-L fed-batch bioreactor, the optimized strain produced PrtA-Q245K/I342D at 4807.5 U/mL, equivalent to 1.5 g/L protein. The combined approach of directed evolution, machine learning-guided mutagenesis, and signal peptide engineering significantly boosted PrtA secretion while maintaining functional integrity. This strategy demonstrates strong potential for scalable industrial production of challenging heterologous proteases.
|
|
Scooped by
mhryu@live.com
March 11, 1:56 PM
|
We therefore propose microbes as simple and scalable model systems to represent a cell that has evolved under selective pressures to adapt to its environment and discuss how microbial genetic diversity can help to improve the performance and benefits of AI models. We argue that, with the help of AI, microbial sequence data from across our planet’s diverse environments can be used to address fundamental biological questions, such as evolution and adaption, how cells make decisions, the relationships and communication between organisms, and how emergent properties arise in biological systems. It would require multi-modal AI models that, in addition to sequence data, also incorporate additional ‘languages’ on environmental and evolutionary processes beyond linear DNA sequences.
|
|
Scooped by
mhryu@live.com
March 11, 1:48 PM
|
Cross-kingdom RNA interference is an emerging concept in plant–pathogen interactions. Here we provide evidence that cross-kingdom RNA interference also occurs in a beneficial plant symbiosis called arbuscular mycorrhiza AMF. The arbuscular mycorrhizal fungus Rhizophagus irregularis transfers small RNAs into plant cells, promoting the colonization of host roots. This finding establishes inter-organismal RNA communication as a new regulatory mechanism of this ancient and widespread symbiosis. The authors provide evidence that symbiotic fungi forming widespread arbuscular mycorrhiza symbioses use cross-kingdom RNA interference to silence plant genes and promote their colonization of host roots.
|
|
Scooped by
mhryu@live.com
March 11, 1:16 PM
|
TnpB is a diverse family of RNA-guided endonucleases associated with prokaryotic transposons. Because of their small size and putative evolutionary relationship to CRISPR–Cas12, TnpB enzymes hold great potential for genome editing. However, most TnpBs lack robust gene-editing activity. Here, we mapped comprehensive sequence–function landscapes of a TnpB ribonucleoprotein using deep mutational scanning and we discovered activating mutations in both the RNA and the protein. Leveraging the protein’s mutational landscape, we constructed a combinatorial library of activating mutations, from which we identified two enhanced TnpB variants. These variants increased editing in human cells, Nicotania benthamiana, pepper and rice. While editing efficiencies varied by target site, engineered variants achieved up to 55% insertion and deletion frequencies (a 50-fold increase over wild type) in N. benthamiana, surpassing ISYmu1 (<7%), AsCas12f-HKRA (<9%) and other compact editors. These findings highlight elements critical for regulating TnpB endonuclease activity and demonstrate latent activity accessible through mutation. TnpB endonucleases are engineered for improved genome editing.
|
|
Scooped by
mhryu@live.com
March 11, 1:07 PM
|
Faecal microbiota transplantation (FMT) is highly effective for recurrent Clostridioides difficile infection but yields inconsistent benefits in chronic indications. As a crude whole-microbiota transplant, FMT contains numerous undefined active components, complicating efforts to ensure treatment predictability and stability. Therefore, we propose Advance Microbiota Transplantation (AMT), a comprehensive, phase-based hypothesis that employs an addition–subtraction strategy throughout the pre-, peri- and post-transplant stages. AMT comprises donor and recipient pre-treatment, procedural optimization and post-transplant adjuvant interventions to mitigate donor variability, ecological resistance, procedural heterogeneity and unstable engraftment. Through a systematic synthesis of current evidence-based FMT research, we explored how the addition–subtraction strategy can be operationalised to shape the AMT concept and define testable, phase-specific levers, thereby providing a foundation for future clinical translation. In parallel, we appraised the reporting quality using the Preferred Reporting Items for Microbiotherapy (PRIM) and identified six persistently under-reported items that limit the interpretability, comparability, and reproducibility of FMT research. This review aims to facilitate the integration of AMT into clinical practice.
|
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
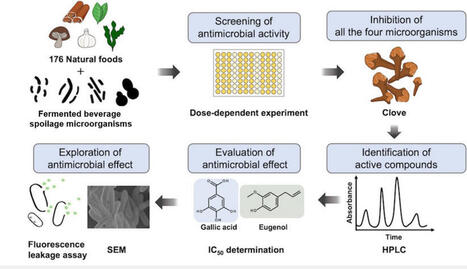
The spoilage of beer significantly threatens the quality and safety of products in the beverage industry. Certain natural foods contain beneficial bioactive components that are considered to be safer than chemical additives. This study aimed to identify extracts of natural foods as potential alternatives to chemical preservatives for controlling beer spoilage microorganisms. Among the two extraction methods applied to 176 natural foods, the extracts of clove alone effectively inhibited the growth of representative beer spoilage microorganisms, specifically Levilactobacillus brevis, Sporolactobacillus vineae, Pectinatus frisingensis, and Saccharomyces cerevisiae var. diastaticus. High-performance liquid chromatography and half-maximal inhibitory concentration analysis revealed that gallic acid and eugenol in cloves were active compounds with antimicrobial properties in vitro that were of a similar order of magnitude to those of commercial preservatives (potassium sorbate and sodium benzoate) under the tested conditions. Scanning electron microscopy observations and a fluorescence leakage assay using 3,3′-dipropylthiadicarbocyanine iodide indicated morphological alterations and membrane perturbation in treated microorganisms, except for the limited effect of gallic acid on S. cerevisiae var. diastaticus. These findings provide insight into the potential role of natural food-derived antimicrobials in controlling beer spoilage microorganisms, pending further validation in real beverage systems.
|
|
Scooped by
mhryu@live.com
March 11, 11:43 PM
|
Anaerobic digestion (AD) is a cornerstone technology for sustainable waste treatment and renewable energy recovery, yet its complex microbe–metabolite interactions remain poorly understood. Here, we combined high-resolution molecular profiling and microbial community sequencing in a three-month study across seven full-scale digesters to resolve dissolved organic matter (DOM) and microbiome dynamics. A total of 28 925 DOM molecules, including a conserved core of 1154 metabolites, were identified. By disentangling metabolic pathways, we observed complex transformation patterns that extend beyond simple substrate breakdown. Molecules within a mass window (183.57–390.81 m/z) exhibited high persistence, strong microbial associations, and distinct transformation trajectories. Within this mass window, microbial community composition and feedstock input, together explained ~30.1%–43.4% of the observed spatiotemporal variation. In each digester, 1260–2108 molecules were closely associated with microbial metabolism, forming 7.77–24.52 microbe–metabolite associations on average. The accumulation and turnover of these microbial metabolites were strongly linked to methane production and system performance, highlighting microbial processing of DOM as a significant factor shaping microbe–metabolite interactions. This perspective emphasizes the importance of microbe–metabolite interplay in AD, providing a conceptual framework for predictive monitoring and optimization of engineered biotechnologies.
|
|
Scooped by
mhryu@live.com
March 11, 11:33 PM
|
Multicellular magnetotactic prokaryotes represent a unique group of obligately marine multicellular bacteria known for their ability to navigate along magnetic field lines thanks to ferrimagnetic nanocrystals. To date, two distinct spherical and ellipsoidal morphotypes have been described, typically ranging from 3 to 6 μm in diameter and comprising approximately 50 cells of the same species. Although widespread in highly reduced marine sediments, they are represented by solely three genera clustering into a monophyletic group within the Desulfobacterota. In this study, we report a third morphotype in reduced sediments of the Mediterranean Sea in Carry-le-Rouet, France, i.e. approximately 30 times more voluminous than any previously described form. Because their large size, we designated these multicellular bacteria as “giant” and explored their cell ultrastructure, ecological niche and physiology using magnetic enrichment and a combination of microscopy techniques and single-consortium genomics. Transmission electron microscopy and confocal microscopy images of several individual consortia revealed that they contain an average of 130 cells, each producing over 100 greigite magnetosomes arranged to optimize the overall magnetic moment. Phylogenomic analyses positioned giant multicellular magnetotactic prokaryotes, together with other morphotypes, in a previously undescribed genus and species within the Candidatus Magnetomoraceae family, named Magnetogigantoglobus mediterraneus. Although genetically divergent with a different ultrastructure, all multicellular magnetotactic prokaryotes seem to rely on sulfate reduction coupled to heterotrophy or autotrophy. We further discuss the significance of these findings in the context of the evolutionary history of multicellularity and magnetotaxis in prokaryotes.
|
|
Scooped by
mhryu@live.com
March 11, 11:25 PM
|
Ice-nucleating proteins (INpros) catalyze ice formation at high subzero temperatures, with major biological and environmental implications. While bacterial INpros have been structurally characterized, their counterparts in other organisms have remained largely unknown. Here, we identify membrane-independent proteins in fungi of the Mortierellaceae family that promote ice formation with high efficiency. These proteins are predicted to adopt β-solenoid folds and multimerize to form extended ice-binding surfaces, exhibiting mechanistic parallels with bacterial INpros. Structural modeling, phylogenetic analysis, and heterologous gene expression leading to ice nucleation in E. coli and Saccharomyces cerevisiae show that the fungal INpros are encoded by orthologs of the bacterial InaZ gene, which was likely acquired by a fungal ancestor through horizontal gene transfer. The discovery of cell-free fungal INpros provides tools for innovative freezing applications and reveals biophysical constraints on ice nucleation across life.
|
|
Scooped by
mhryu@live.com
March 11, 5:18 PM
|
Nitrogen (N) and phosphorus (P) are indispensable macronutrients for crop growth and productivity; however, their excessive application in agriculture has caused severe environmental degradation. Enhancing crop N-use efficiency (NUE) and P-use efficiency (PUE) is a critical strategy to reconcile high productivity with sustainability. In this review, we systematically synthesize recent advances in the genetic basis of NUE and PUE in crops, focusing on key traits and their associated signaling networks. We summarize the identification of N/P-efficiency genes and explore how natural variations in these genes correlate with soil nutrient availability, revealing adaptive patterns from crop domestication. Given the distinct biogeochemical behaviors of N and P, we propose tailored strategies that leverage nutrient-specific traits to optimize environment–resource coordination and yield–quality balance. Finally, we discuss strategies for developing future crop cultivars with enhanced NUE or PUE to advance sustainable agriculture.
|
|
Scooped by
mhryu@live.com
March 11, 3:21 PM
|
Genome-editing technologies that use recombinases to insert kilobase-scale DNA sequences into mammalian genomes canonically require large double-stranded DNA (dsDNA) donors. However, dsDNA molecules evoke problematic and toxic innate immune responses, limiting integration efficiencies and generally constraining applicability to ex vivo or immune-deficient contexts. By harnessing mechanisms of integrative prokaryotic viruses and mobile genetic elements, here we demonstrate that recombinases are compatible with immune evasive circular single-stranded DNA molecules optimally bearing a partial-duplex region that reconstitutes the recombinase recognition sequence. This approach, which we term integration through nucleus-synthesized template addition of large lengths (INSTALL), is compatible with diverse protein and RNA-guided recombinases for high-fidelity kilobase-scale human genome writing. INSTALL minimizes innate immune responses in primary human cells and in mice, improving recombinase-mediated integration efficiencies and supporting systemic in vivo non-viral DNA delivery by substantially increasing tolerability and broadening the dosing range compared with lipid nanoparticle-delivered dsDNA molecules. Together, INSTALL overcomes fundamental challenges for DNA delivery and integration methods by synergizing immune-stealth nucleic acids with recombinases to enable kilobase-scale integration strategies without viral vectors. INSTALL overcomes fundamental challenges for DNA delivery and integration methods by synergizing immune-stealth nucleic acids with recombinases to enable kilobase-scale integration strategies without viral vectors.
|
|
Scooped by
mhryu@live.com
March 11, 3:14 PM
|
Engineered cell therapies are transforming precision medicine by enabling real-time, context-responsive interventions that act upon disease-specific cues. Inspired by the success of CAR-T cells in oncology, next-generation platforms are being developed using diverse immune cells and stem cells to address a broader spectrum of diseases. These living therapeutics harness synthetic gene circuits to induce targeted cytotoxicity, to modulate the secretion of effector proteins or to coordinate both functions in response to endogenous signals or externally delivered molecular and physical triggers. Ex vivo engineering of autologous cells remains the norm, but challenges in scalability, cost and accessibility are fuelling efforts towards allogeneic products and in vivo reprogramming. Advances in targeted delivery — using viral vectors, mRNA-loaded nanoparticles and virus-like particles — are expanding the toolkit for direct programming of cells within the body. This Review discusses emerging strategies for engineering human cells with therapeutic functions, highlighting modular control systems, delivery innovations and the translational hurdles that lie ahead. In this Review, Teixeira et al. discuss emerging strategies for developing and improving engineered-cell therapies. They outline progress from ex vivo engineered autologous cells to in vivo reprogramming, advances in delivery systems and the remaining translational barriers.
|
|
Scooped by
mhryu@live.com
March 11, 2:49 PM
|
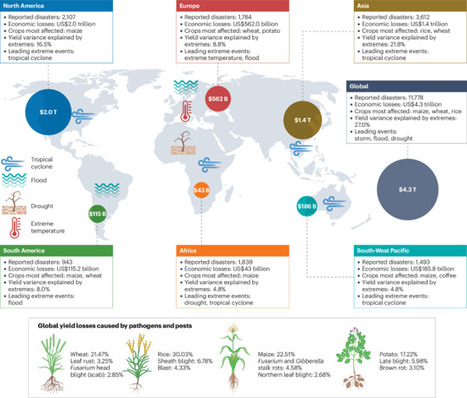
Plant diseases pose a great risk to global food security, and recent research indicates that pathogen pressures on plant productivity will substantially increase under ongoing climate change (that is, increasing CO2 levels and global warming). However, our mechanistic and predictive knowledge of the impacts of climate extremes, such as heatwaves and prolonged droughts, and their interaction with other climatic factors, on plant pathogens, hosts and microbiomes, remains largely unknown. This is an important knowledge gap that limits our ability to develop effective strategies to mitigate the socioeconomic impacts of climate change-induced plant disease outbreaks. This Review examines the impacts of key climate extremes on soil-borne pathogens, plant microbiomes and host physiology that ultimately determine disease outcomes. We explore evidence that suggests that the responses of pathogen–host–microbiome interactions to climate extremes may differ in many ways from those to long-term climate change. Climate extremes may increase the virulence and distribution of many pathogens, suppress certain plant immune responses, and weaken the core functions of host microbiomes within the disease triangle, thereby facilitating disease outbreaks. We propose an integrated pathway for harnessing microbiomes to address the critical challenges posed by climate extremes. These insights offer new approaches to mitigate disease risks by harnessing microbiomes and metabolites under climate extremes, with the potential to support climate-resilient and sustainable agricultural and natural ecosystems. In this Review, Singh BK and colleagues discuss the impacts of key climate extremes on soil-borne pathogens, plant microbiomes and host physiology that ultimately determine disease outcomes, as well as the eco-evolutionary mechanisms by which pathogens, hosts and microbiomes may adapt to climate extremes. Finally, they propose an integrated pathway for harnessing microbiomes to address the critical challenges posed by climate extremes.
|
|
Scooped by
mhryu@live.com
March 11, 2:40 PM
|
Genome-scale targeted CRISPR libraries for forward genetic screens in plants are powerful tools for functional analysis, but they suffer from limited spatial control, single sgRNA design, and poor handling of genetic redundancy. We develop multiplexed CRISPR libraries in which each construct contains two sgRNAs that simultaneously target multiple members of a gene family. The libraries can also function at the cell-type-specific and tissue levels. A double-barcoding strategy enables efficient tracking and identification of sgRNA combinations at the plant level without individually sequencing each line. Using this platform, we generate over 1,000 Arabidopsis lines that express sgRNAs targeting 707 transporter genes across 114 gene families involved in nutrient uptake. The multiplexed design increases gene coverage and editing efficiency, underscoring its improved targeting capability to reveal hidden phenotypes. This toolbox provides a scalable resource for multi-targeted genome editing and spatially precise forward genetic screens in plants.
|
|
Scooped by
mhryu@live.com
March 11, 1:52 PM
|
Artificial intelligence has rapidly entered the field of life sciences, including vaccine development. Here, we introduce ‘reverse vaccinology 3.0’, a transformative approach that could revolutionize the field of vaccinology and contribute to addressing global health challenges. Andreano, McLellan and Rappuoli introduce ‘reverse vaccinology 3.0’, a transformative approach that could revolutionize the field of vaccinology and contribute to addressing global health challenges.
|
|
Scooped by
mhryu@live.com
March 11, 1:40 PM
|
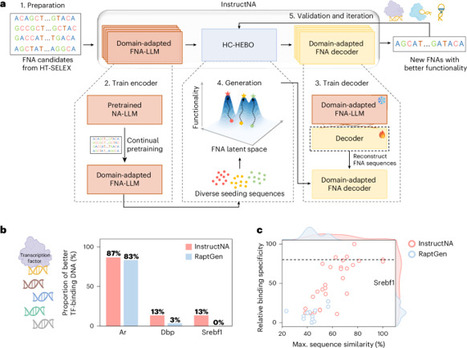
Functional nucleic acids (FNAs) are essential elements for designing advanced molecular tools, yet their de novo design faces challenges due to the vast sequence space and inefficiency of experimental screening methods. Nucleic acid large language models (NA-LLMs) offer new opportunities for FNA design, but their generative capability remains underexplored. Here we introduce InstructNA, a framework leveraging NA-LLMs and high-throughput systematic evolution of ligands by exponential enrichment (HT-SELEX) to guide de novo design of FNAs without relying on structural information. InstructNA encodes semantically rich FNA representations and robustly decodes FNA sequences, enabling the generation of various types of FNA such as transcription factor-binding DNA and protein-binding aptamers with enhanced functionality and high sequence diversity. Compared with the traditional HT-SELEX, InstructNA generates 100% and 200% more strong aptamer binders for two protein targets, with a sequence similarity to the original HT-SELEX aptamers as low as 38%. These results underscore the efficacy and robustness of InstructNA, demonstrating its potential for FNA design. InstructNA leverages nucleic acid large language models with HT-SELEX for de novo generation of functional nucleic acids, exhibiting high efficiency and general applicability in designing aptamers for various targets.
|
|
Scooped by
mhryu@live.com
March 11, 1:13 PM
|
The transcriptional interference model suggests that RNA polymerases elongating through overlapping transcription units mutually inhibit transcription and disrupt associated cis-regulatory elements. As a longstanding fundamental concept of gene regulation, the idea of reciprocal inhibition between sense and antisense transcription has been supported by a significant body of research. However, despite the model’s biophysical plausibility and historical significance, evidence from large-scale transcriptome studies raises questions about its universal applicability. In particular, the new data indicate that a measurable influence of transcriptional interference is absent from the majority of loci with overlapping transcription. Here we highlight key aspects of overlapping transcription and propose potential solutions to this emerging puzzle. Gaining a better understanding of the molecular mechanisms that render loci sensitive or resistant to interference could lead to groundbreaking insights into the biology of gene regulation. The transcriptional interference model has been instrumental in shaping our understanding of gene regulation. However, given that its effects are largely absent from genome-wide data, this Perspective reexamines the underlying mechanisms and suggests how they may be resolved at most loci.
|






























Based on screening the hydrolysis zone on milk agar plates