Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:14 PM
|
Metal ions play critical structural, regulatory, and enzymatic roles in proteins, making their binding essential for biological processes. Experimental identification of metal-binding sites is resource-intensive and limited in scalability. Recent advances in protein language models have transformed computational predictions, yet current tools do not address how residue-level metal-binding probabilities change upon mutation. To fill this gap, mCSM-metal leverages embeddings from ESMBind with our graph-based structural signatures to accurately predict the effects of single or multiple point mutations on the binding of seven essential ions (Zn2+, Ca2+, Mg2+, Mn2+, Fe3+, Co2+, Cu2+). Our model achieves accuracies, F1-scores, and Matthews Correlation Coefficient values up to 0.97, 0.97, and 0.95, outperforming other approaches. The webserver provides an interactive platform to assess and visualize local and long-range impacts of mutations on metal-ion binding, offering new avenues for applications in structural biology, disease modelling, and protein engineering. The web application is freely available at: https://biosig.lab.uq.edu.au/mcsm_metal/.
|
|
Scooped by
mhryu@live.com
Today, 12:03 PM
|
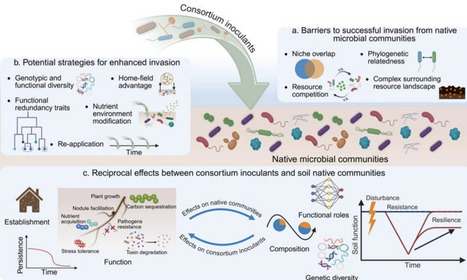
Synthetic microbial consortium inoculants are emerging nature-based solutions for promoting sustainable agriculture and mitigating environmental challenges. However, despite promising results in simpler lab-scale trials, many inoculants fail to establish or perform satisfactorily in field conditions. One most critical yet least understood factor influencing inoculant effectiveness is the complex microbial interactions, both within consortium inoculants (“within-community” interactions) and between consortium inoculants and native soil communities (“cross-community” interactions). Here, we first discuss major negative and positive “within-community” interactions and highlight the importance to design consortium inoculants with positive interactions for improved stability and functionality. We then examine the bidirectional “cross-community” interactions once introducing consortium inoculants to soils. Soil native communities often create strong resistance to the invasion of inoculants. We discuss major drivers controlling the invasibility of native communities and various strategies increasing the invasiveness of consortium inoculants. We then discuss how consortium inoculants can reshape native communities, with implications for long-term ecosystem resilience and functioning. We propose future research efforts including advancing strategies for harnessing natural species from relatively untapped soil reservoirs and using high-throughput interaction profiling with computational tools to build compatible synthetic consortia with desirable functions; leveraging positive interactions and prebiotics to facilitate inoculant establishment; and assessing fully soil functional resilience over longer terms, including recognizing the importance of rare keystone taxa. By integrating with ecological theory, this review provides a comprehensive insight into microbial interactions to advance the design, application, and monitoring of synthetic consortium inoculants for enhancing soil health and ecosystem sustainability.
|
|
Scooped by
mhryu@live.com
Today, 11:55 AM
|
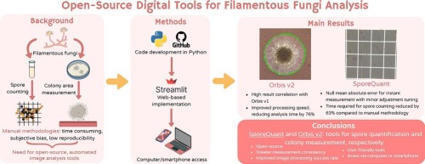
Reproducibility and standardization of methods are key approaches to microbiological research. However, laboratories differ widely in equipment, protocols, and operator expertise. These disparities are especially pronounced in fungal biology, since accurate spore counting and precise measurements of colony growth remain vulnerable to subjective bias and inconsistent technique. To standardize these foundational tasks, we developed two free, open-source image-analysis tools – SporeQuant and Orbis v2 – that automate hemocytometer spore counting and colony-area measurement, respectively. SporeQuant applies image thresholding and blob detection to identify individual spores and compute their concentration, while Orbis v2 segments colonies and calculates colony areas through thresholding and contour detection. Compared with manual analysis, SporeQuant and Orbis v2 markedly reduce analysis time by 63% and 76%, respectively, while achieving high measurement reliability, with MAE = 0 for SporeQuant and R2 > 0.975 for Orbis v2. These tools reduce user-to-user variability, and achieve comparable or improved accuracy versus previously established methods. Furthermore, both tools can be accessed through a web page on both a computer or smartphone, requiring no specialized hardware, software, or prior knowledge. They provide accessible solutions for low-budget and high-throughput laboratories alike, enabling more consistent, scalable analysis of fungal growth and morphology in research and industrial settings.
|
|
Scooped by
mhryu@live.com
Today, 11:38 AM
|
Reducing dependence on synthetic nitrogen fertilizer requires biologically grounded alternatives. Symbiotic nitrogen fixation supplies fixed nitrogen but is restricted to a narrow angiosperm clade, limiting direct deployment in most major nonleguminous crops. We synthesize how telomere-to-telomere genomes and pangenomes expose structural and regulatory variants for nodulation; how single-cell and spatial transcriptomics resolve stage-specific cell states and division of labor; and how epigenomic and 3D genome maps reveal principles of regulatory control for infection, organogenesis, and fixation. Extending to actinorhizal symbioses tests single- versus multiple-origin models. We present an artificial intelligence-guided roadmap that integrates sequence, chromatin accessibility, and expression data to prioritize regulatory elements, propose compact edit sets, and guide cell type-specific deployment in nonleguminous crops, advancing from descriptive catalogs to testable models and iterative validation.
|
|
Scooped by
mhryu@live.com
Today, 10:32 AM
|
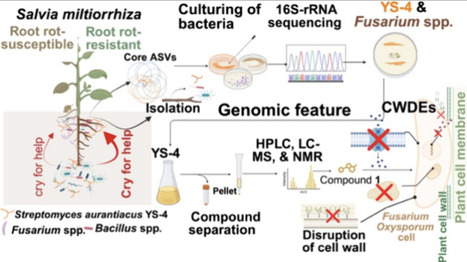
Microbiome-guided crop protection increasingly emphasizes the chemical basis of disease suppression. Here, we identify Streptomyces aurantiacus YS-4, a rhizosphere-enriched actinobacterium selectively recruited by resistant cultivars of Salvia miltiorrhiza, as a producer of metacycloprodigiosin, a previously reported secondary metabolite that is reported for the first time in this strain and further investigated for its role in suppressing Fusarium oxysporum-induced root rot in S. miltiorrhiza. Metacycloprodigiosin inhibited Fusarium oxysporum growth by 81.66% at 200 μg mL–1 and induced extensive cellular and transcriptional changes related to membrane function and virulence-associated pathways. Transmission electron microscopy confirmed severe hyphal damage, while transcriptomic profiling revealed broad downregulation of virulence-associated genes. Pot experiments demonstrated that YS-4 application alleviated root rot symptoms and enhanced plant biomass. Collectively, these findings establish a direct link between metabolite chemistry and pathogen suppression, advancing molecular understanding of plant–fungal interactions and highlighting metacycloprodigiosin as a promising biocontrol agent for sustainable management of crops.
|
|
Scooped by
mhryu@live.com
Today, 10:24 AM
|
Intensity and duration of biological signals encode a few pathways to direct diverse cellular behaviors, yet quantifying these features in single cells remains difficult. To address this challenge, we developed INSCRIBE, which uses a CRISPR base editor to mutate genomic targets at rates proportional to signaling activity. Edits are recovered at the endpoint through a new ratiometric readout strategy from images of two fluorescence channels. We engineered human cells to record WNT and BMP activity. Following defined exogenous stimulations, INSCRIBE accurately recovered signal intensity in dose–response experiments and exposure duration in time-course experiments. Applying INSCRIBE revealed a persistent memory in the BMP pathway, where progeny of high-responding cells remained more sensitive to subsequent BMP stimulation for up to 3 weeks. Together, our results establish a scalable platform for genetic recording and in situ readout of signaling activity in single cells, advancing quantitative analysis of cell–cell communication during development and disease. Hao et al. developed a CRISPR-based system that records cell signaling activity as DNA edits readable by imaging, enabling single-cell reconstruction of signal strength and revealing long-lasting memory in the BMP pathway.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
Emerging fungal pathogens represent a concerning threat to both global health and food security. In this study, we aimed to address our rising vulnerability to fungal pathogens through the development of the Fung-AI pipeline: an AI/ML-driven approach for antifungal discovery. A generative adversarial network (GAN) was trained to generate novel candidate antifungal peptide sequences. Next, in silico antifungal and hemolytic classifiers were built to further prioritize AI-generated peptides for experimental validation. From a pool of ~10,000 candidates, thirteen peptides were selected for testing over two-stages of experimentation. Five peptides were found to display mild antifungal activity against the wheat pathogen, Fusarium graminearum, with minimal inhibitory concentrations (MICs) ranging from 250 µg/mL to 500 µg/mL. Four of the five peptides also showed activity against the human pathogen, Candida albicans (MIC: 500 µg/mL). Two of our AI-generated antifungal peptides additionally demonstrated low cytotoxicity in HepG2 human liver carcinoma cells (LC50 > 704.2 µg/mL) indicating that they may be useful as scaffolds for future optimization for therapeutic applications. None of our peptides were found to considerably inhibit the emerging pathogen C. auris, suggesting the need for pathogen-specific down-selection of candidate peptides. Overall, we present a proof-of-principle, generative-AI-based approach for the rapid design of de novo antifungal peptides.
|
|
Scooped by
mhryu@live.com
Today, 1:08 AM
|
Phages can modify host cell physiology to thwart competitors. The Pseudomonas aeruginosa-specific phage DMS3 encodes Aqs1, a protein inhibitor of type IV pilus (T4P) function to prevent host cell recognition by other phages that leverage these filaments for infection. Aqs1 disrupts T4P by binding to the hexameric ATPase PilB, required to power pilus filament extension, though several mechanistic details remain unclear. We show that Aqs1 has broad-spectrum activity and can disrupt T4P function in a variety of Gram-negative bacteria. This protein inhibits PilB by binding to a solvent-exposed hydrophobic patch on the N2-domain, distal to the active site. Binding destabilizes the hexamer, preventing PilB accumulation at T4P machines. Aqs1 likely disrupts PilB oligomerization by displacing a flexible linker segment between the PilB N1- and N2-domains required for inter-subunit contact. Together, the Aqs1 mode of action provides a design template for broad-spectrum inhibitors of diverse bacterial virulence factors.
|
|
Scooped by
mhryu@live.com
Today, 12:10 AM
|
Most non-model Vibrio species lack the genetic tools needed for targeted mutagenesis, which limits the ability to functionally characterize newly identified pathways. To address this challenge, we present here efficient, robust methods for genetically manipulating Vibrio species that rely on RecA-mediated homologous recombination and two well-characterized counterselection methods, galactokinase (galK) 2-Deoxy-D-galactose (DOG-2) toxicity and the rpsLR/rpsLS streptomycin susceptibility system, both of which are active across a broad range of Vibrio species. We further characterized two genus-specific conserved promoters capable of driving high-level ectopic expression across all tested species. These promoters were incorporated into two broadly applicable, conjugatively transferable suicide backbones designed to facilitate double homologous recombination. Using these systems, we successfully disrupted polar flagellar motility in multiple Vibrio species and introduced extensive modifications to both flagellar and secretory pathways in V. diazotrophicus. Notably, although the galK system exhibited broader applicability, the rpsL system proved to be more efficient in cases where a streptomycin resistant strain could be generated. We also developed two mobilizable replicative backbones that express pH-stable fluorescent proteins for use within the genus. Collectively, these tools expand the genetic toolkit available for both gene disruption and heterologous gene expression in non-model members of the Vibrionaceae.
|
|
Scooped by
mhryu@live.com
March 10, 11:07 PM
|
Whole-cell biosensors (WCBs) capable of sensitively detecting trace amounts of analytes hold great potential for in situ detection of pollutants, toxins, or synthetic products. As the terminal signal actuator, the reporter gene directly influences the ultimate sensitivity of WCBs. Although fluorescent proteins (FPs) have been widely used as reporters, their reporting sensitivity is generally lower than that of enzymatic reporters, which often limits the sensitivity and response speed of FP-based sensors in practical applications. Here, we developed an ultrasensitive FP reporter via a noninvasive N-terminal peptide fusion strategy. By adding an N-terminal decapeptide obtained from a high-throughput screening, we constructed an NGFP4 variant that retains the inherent advantages of sfGFP while exhibiting superior reporter gene characteristics, such as rapid expression and robust intracellular stability. These properties enhanced the single-cell fluorescence intensity of NGFP4 by 6.4- to 28-fold in four typical microbial hosts, including E. coli (28-fold), Bacillus subtilis (15.5-fold), Pichia pastoris (9.1-fold), and Saccharomyces cerevisiae (6.4-fold). When applied to WCBs, the NGFP4 reporter greatly shortened the detection time to 1 h for salicylic acid (LOD of 0.36 μM) and 2-chlorobiphenyl (LOD of 18.2 μM), representing the fastest detection time for such sensors. Therefore, our work provides a cross-species compatible FP reporter that enables sensitive detection of microbial cell-based biosensors and other bioanalytical systems, facilitating their field deployment with minimal genetic manipulation and shorter detection time.
|
|
Scooped by
mhryu@live.com
March 10, 10:56 PM
|
CRISPR-associated proteins (Cas) are central to gene editing, forming nuclease complexes with guide RNA to enable precise genome modification. Among numerous Cas variants, Cas9 and Cas12a are the most extensively studied. While much is known about the genomic substrates for these enzymes, less is known about the determinants of the DNA cleavage activity. Wild-type Cas12a exhibits higher intrinsic specificity than Cas9, minimizing off-target activity, but lower overall potency. Recent protein engineering has sought to improve both parameters. Here, we shed light on the structural and mechanistic basis by which an engineered AsCas12a variant achieves high potency while retaining its hallmark specificity. We show that reduced protein–DNA interactions facilitate more rapid R-loop formation, thereby enhancing cleavage activity. These results provide mechanistic insight into Cas12a function and highlight strategies for designing genome-editing nucleases with optimal balance between efficiency and specificity. Engineered AsCas12a increases gene editing potency, retaining high specificity. Potency increases by reducing protein-DNA interactions, mediating faster R-loop formation. This provides insight into design of efficient and precise CRISPR nucleases.
|
|
Scooped by
mhryu@live.com
March 10, 10:45 PM
|
Bacteria play fundamental roles in ecosystems, human health, and biotechnology. Although bacterial genome sequencing data have accumulated rapidly over the past decade, the metabolic and ecological functions of most sequenced bacteria remain poorly understood, apart from a few well-studied taxa and traits. Establishing a general framework that comprehensively captures the relationship between bacterial genomes and the diverse biological functions they encode remains a major challenge, as this task requires embedding individual genes within their broader genomic context and modeling the combined effects of gene interactions across complex biological pathways and networks. The difficulty is further compounded by the limited functional annotations available for most bacterial genomes. Here, we introduce BacPT, a bacterial proteome foundation model trained on tens of thousands of complete genomes spanning diverse taxa. BacPT captures both local and genome-wide information, enabling the generation of contextualized gene embeddings and functionally rich representations at the whole organism level. We demonstrate the utility of BacPT across diverse prediction tasks spanning multiple biological scales. BacPT embeddings improve the prediction of enzyme activities, biosynthetic gene clusters BGC, metabolic traits, and ecological interaction outcomes. Our results highlight that unsupervised deep learning applied at the scale of entire proteomes provides a powerful approach for characterizing gene interactions and mapping functional landscapes for bacteria.
|
|
Scooped by
mhryu@live.com
March 10, 5:05 PM
|
Advanced biological imaging analysis platforms such as Activity Quantification and Analysis (AQuA2) enable accurate spatiotemporal activity analysis across diverse cell populations within many species. These tools are increasingly important for investigating cellular signaling dynamics and behavior. However, despite advances in the accuracy and species capability of AQuA2, it remains computationally demanding for analysis of long time-series datasets and requires all users to maintain a MATLAB license, which may limit accessibility and large-scale deployment. To address these limitations, we have designed and made available AQuA2-Cloud, a portable software stack and web platform developed as an improvement and further evolution of AQuA2. This container-deployable system permits multi-user cloud-based high accuracy activity quantification with intuitive workflows, export of analysis data and project files, and comparable processing times. The platform offers integrated features such as in-browser analysis control interfaces, asynchronous program state control, multiple users and user management, support for unreliable connections, file uploading and downloading via web browsers and File Transfer Protocol, and centralized organization of analysis output. AQuA2-Cloud constitutes a cloud-native solution for laboratories or research groups seeking to centralize analysis of spatiotemporal biological imaging datasets while reducing software installation and licensing barriers for end users. The platform enables researchers with minimal technical expertise to perform advanced bioimaging analysis through standard web browsers while maintaining the analytical capabilities of AQuA2. AQuA2-Cloud source code, deployment procedures, and documentation are freely available at (https://github.com/yu-lab-vt/AQuA2-Cloud).
|
|
|
Scooped by
mhryu@live.com
Today, 12:13 PM
|
Type III secretion (T3SS) systems assemble bacterial nanomachines, including the flagellum and virulence-associated injectisomes, by exporting distinct classes of substrates in a defined temporal order. In both systems, completion of an early assembly intermediate triggers an irreversible switch from early to late substrate secretion. In the flagellar system, this switch is controlled by the secreted molecular ruler FliK acting on the core T3S component FlhB, but the molecular mechanism governing this transition has remained unclear. Here we show that removal of two components, Fluke and the cleaved C-terminal domain of FlhB (FlhBCCD), locks the secretion apparatus in a constitutive late secretion state. In these mutants, secretion specificity no longer requires completion of the hook-basal body or the FliK ruler, indicating that Fluke and FlhBCCD function to maintain the apparatus in early secretion mode. Consistent with this model, synchronized flagellar gene expression experiments reveal that FlhBCCD is retained during early assembly and is lost coincident with hook-basal body completion and activation of σ28-dependent late gene expression of flagellin and chemosensory genes. Structural modeling of the FliK C-terminal switch domain and FlhBCCD supports a mechanism in which secretion of FliK promotes destabilization and ejection of FlhBCCD from the secretion apparatus. Disruption of a folded region within FliK switch domain uncouples secretion from switching, indicating that the timing of FliK unfolding during secretion is critical for activation of the specificity switch. These findings show that secretion specificity switching is driven by FliK-dependent removal of inhibitory components, rather than passive sensing of assembly completion.
|
|
Scooped by
mhryu@live.com
Today, 11:59 AM
|
Biohybrid microrobots integrate biological components with synthetic structures to navigate complex biological environments, for example, for the delivery of drugs, microsurgery and in vivo diagnostics. In this Review, we propose a biophysics-informed design framework for biohybrid microrobots by connecting biophysical principles with biohybrid solutions. We first identify the biophysical constraints imposed by the human body that limit microrobot integrity, locomotion, navigation and functionality. We then examine the biophysical mechanisms through which biological cells, microorganisms and their derivatives adapt to these challenges, and explore how these can be utilized to improve the performance of microrobots. Building on these insights, we describe how biohybrid microrobots translate biophysical strategies into engineering solutions across four design domains: deformation, actuation, navigation and programming. Finally, we discuss persisting in vivo challenges, key considerations for clinical translation and future developments. By articulating design logics that span biological and synthetic domains, this framework provides a functional definition of biohybrid microrobots and offers a shared language for researchers across disciplines. Biohybrid microrobots combine biological and synthetic components to navigate complex in vivo environments for applications such as drug delivery, microsurgery and diagnostics. This Review introduces a biophysics-informed design framework for biohybrid microrobots that translates natural adaptive strategies into engineering solutions across deformation, actuation, navigation and programming.
|
|
Scooped by
mhryu@live.com
Today, 11:49 AM
|
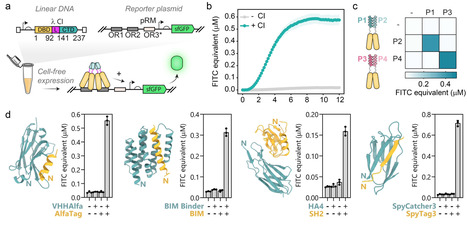
Protein binders that detect, activate, inhibit, or otherwise modulate their targets are pivotal for biomedical applications. With the increasing accuracy and accessibility of de novo protein design, faster and cheaper experimental screening methods would democratize and accelerate the identification of high-affinity binders. Here we present Cell-Free Two-Hybrid (CF2H), a rapid and sensitive method for detecting high-affinity protein-protein interactions (PPI) that does not require cloning, protein purification nor high-end laboratory equipment. CF2H uses a dimerization-activated DNA binding domain (DBD) fused to prey and bait proteins to trigger transcription upon protein-protein interaction. We demonstrate that CF2H enables the detection of interactions between various types of target and binder proteins such as single-domain antibodies, DARPins, and de novo designed binders. We benchmark CF2H as a screening platform by validating previously reported binders for Mdm2 and discovering high-affinity binders targeting the checkpoint inhibitor PD-L1 in less than 24 hours. Finally, we show that CF2H can be used to characterize small-molecule modulators of PPI and detect protein biomarkers, opening the door for a new class of cell-free biosensors. Faster and cheaper experimental screening methods will democratize and accelerate the identification of high-affinity binders with biomedical applications. Here the authors present Cell-Free Two-Hybrid (CF2H), a rapid and sensitive method for detecting high-affinity protein-protein interactions.
|
|
Scooped by
mhryu@live.com
Today, 11:04 AM
|
AlphaFold, a groundbreaking artificial intelligence model developed by DeepMind, has transformed the field of structural biology by predicting protein structures with unprecedented accuracy. Despite its widespread recognition and application across academia and industry, comprehensive reviews detailing AlphaFold's unexpected applications within the molecular sciences remain scarce. In this review, we critically examine AlphaFold's emerging roles across diverse molecular scientific disciplines. Specifically, we highlight its applications in enzyme engineering and drug development, nucleic acid modeling and vaccine design, the development of protein-based materials and targeted drug delivery systems, and modeling of complex systems and biological networks. To conclude, the review outlines potential future developments and enduring challenges within the application of AlphaFold to molecular sciences. Overall, this review aims to systematically analyze the most recent advances; explore novel interdisciplinary applications of AlphaFold within the realms of biology, chemistry, and materials science; and offer insights into future directions for research and application.
|
|
Scooped by
mhryu@live.com
Today, 10:29 AM
|
One-carbon (C1) substrates are promising feedstocks for microbial bioproduction. Methylobacterium, known for its exceptional C1 utilization capacity, has emerged as a model microbial chassis for sustainable biomanufacturing. In this review, we first outline the C1 assimilation pathways in Methylobacterium and underscore its potential for producing valuable native metabolites. Furthermore, we then survey the genetic tools available for engineering this genus, including plasmid-based methods, transposon mutagenesis, homologous recombination, and CRISPR/Cas systems. Notably, recent advances in metabolic engineering have significantly expanded its biosynthetic scope, enabling the biosynthesis of diverse non-native compounds. Beyond its biomanufacturing potential, Methylobacterium also serves as a versatile plant growth-promoting bacterium, enhancing plant health and productivity through hormone synthesis, nutrient mobilization, stress mitigation, and induced systemic resistance. Collectively, this work highlights the dual potential of Methylobacterium as a sustainable microbial cell factory for biomanufacturing and a beneficial bioinoculant for agriculture.
|
|
Scooped by
mhryu@live.com
Today, 10:03 AM
|
The in-situ upcycling of decentralized methane and nitrogen gas (N2)-derived ammonia via methanotrophic bacteria is highly attractive. However, the toxic intermediate generated from ammonia oxidation significantly inhibits cell growth, thereby hindering efficient bioproduction. Herein, by integrating transcriptomic analysis, we develop rational metabolic engineering strategies and an optimized fed-batch fermentation to enhance ammonia utilization in a methanotrophic bacterium of Methylotuvimicrobium sanxanigenens. The modified M. sanxanigenens overexpressing hydroxylamine reductase efficiently co-assimilates methane and ammonia for cell protein synthesis, with an 18-fold increase in productivity. The resulting methanotrophic cell protein (MCP) not only exhibits an ideal essential amino acid profile but also contains bioactive nutrients, including polysaccharides and peptides. Oral administration of this nutritional MCP significantly ameliorates colitis symptoms in male mice by attenuating inflammatory progression and restoring the intestinal barrier. Moreover, MCP treatment maintains gut microbiota homeostasis and promotes the excretion of beneficial metabolites, thereby protecting the intestinal microenvironment. Hence, this study provides a promising biological approach for the local bio-valorization of decentralized CH4 and air into functional feed additives. This biotechnology not only facilitates advancements in developing carbon-negative gas-to-value pathways but also drives green transformations in animal husbandry by reducing the use of antibiotics and vaccines. Methanotrophic growth on methane and N2-derived ammonia is inhibited by a toxic intermediate, hydroxylamine, generated via ammonia oxidation. Here the authors ameliorate this toxicity through genetically engineering M. sanxanigenens and feed the biomass to mice to reduce colitis symptoms.
|
|
Scooped by
mhryu@live.com
Today, 1:12 AM
|
The precise mapping between chemical transformations and enzymatic catalysts underpins the complexity of metabolic networks. Conventional discovery methods, tethered to sequence homology or structural alignment, are inherently blind to new reactions. Here we present VenusRXN, a multimodal deep learning framework that shatters this limitation by enabling reaction-conditioned enzyme discovery. By seamlessly unifying a pre-trained reaction encoder with a protein language model, VenusRXN achieves a fine-grained, high-dimensional alignment of chemical and biological representations. On benchmarks to discover enzymes which catalyze reactions not seen in the training dataset, it surpasses state-of-the-art baselines with a top-20 retrieval hit rate of 76.5%. Most critically, we demonstrate VenusRXN's capabilities in a zero-shot discovery. As verified by the wet-lab experiments, it successfully identified enzymes to catalyze the chemical reactions never reported, including the one to catalyze the synthesis route for a type 2 diabetes drug intermediate using a non-natural substrate. With surprising precision, the model pinpointed active candidates within the top 10 sequences directly from a global search space of over 300 million proteins, which can hardly be achieved by structure-based enzyme discovery algorithm. This work signals a definitive paradigm shift, establishing the chemical reaction itself, rather than homology, as the primary functional descriptor for the de novo discovery of biocatalysts.
|
|
Scooped by
mhryu@live.com
Today, 1:05 AM
|
Bacteriophages are ubiquitous in nature, but relatively few have been isolated and characterized compared to the number of bacterial strains. Phage biotechnology applications benefit from a diverse library of isolated phages to kill or transfer genetic material to a bacterium of interest. However, scaling phage discovery for diverse bacterial hosts can be time consuming and costly. We developed an approach to capture novel phages for multiple bacteria strains in parallel from an environmental sample using commercially available 0.2-micron filter plates. Using this High-throughput Phage Isolation Platform (HtPIP), we isolated twelve novel phages spanning nine diverse bacterial host genera. Eleven of the isolated phages define new phage species with nine also defining new genera. We show the HtPIP can discover both DNA and RNA phages; including a Tectiviridae infecting Pseudomonas putida mt-2 and a Leviviricetes infecting a Microbacterium isolate, which represents the first cultured RNA phage infecting a host outside of proteobacteria. Using a metagenomic approach, we demonstrate that the HtPIP captures a higher proportion of novel phages compared to traditional low-throughput methods.
|
|
Scooped by
mhryu@live.com
Today, 12:06 AM
|
Prime editing (PE) enables the precise installation of targeted insertions, deletions, and all possible base-to-base conversions without introducing double-strand breaks or donor DNA templates. However, its efficiency remains highly variable across genomic contexts. To address this, multi-faceted optimization strategies have been developed: protein engineering has yielded editor variants with enhanced reverse transcriptase activity and stability; structural refinements to pegRNA design improve its functional integrity and resistance to degradation; regulation of the PE-Flap-mismatch repair (MMR) process favors the retention of desired edits; and the development of protospacer adjacent motif (PAM)-relaxed Cas variants dramatically expands targetable sites. This review systematically consolidates these advances, illustrating how the integration of structural, mechanistic and targeting enhancements is overcoming fundamental bottlenecks. Together, these developments establish PE as a versatile and efficient system for precision genome engineering, paving the way for its reliable application in diverse biological settings.
|
|
Scooped by
mhryu@live.com
March 10, 11:01 PM
|
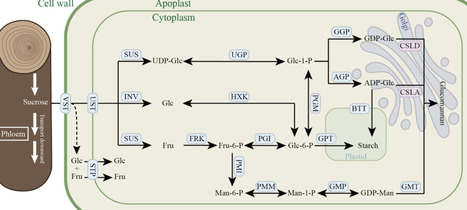
Saccharides, a class of essential organic compounds, are ubiquitously found in nature and play a critical role in vital biological processes. They serve as the primary energy source for all living organisms, supporting life functions. In recent years, the synthesis of saccharides via synthetic biology has gained significant attention, with plant systems emerging as a promising alternative to traditional microbial hosts. Plant chassis offer a unique platform by combining photosynthetic carbon fixation with native saccharide biosynthesis and metabolism, enabling sustainable and potentially large-scale saccharide production. This review highlights the advancements in key technologies for saccharide production using plant chassis, summarizes the current research status of plant-based saccharide production across various structural forms, and discusses the technical challenges and strategies for system optimization. The aim is to provide valuable insights for the development of synthetic biology and uncover the commercial potential of plant chassis in saccharide biosynthesis.
|
|
Scooped by
mhryu@live.com
March 10, 10:49 PM
|
Predicting gene function is a pivotal and challenging step in genomic and metagenomic data analysis. Current automatic annotation tools typically rely on the single most similar sequence from the query database. The sparsity of data per annotation makes it challenging to confidently assign gene function for underrepresented genes. Here, we present a contrastive learning framework for functional annotation. FAMUS (Functional Annotation Method Using Supervised contrastive learning) compares query sequences to profile Hidden Markov Model databases and transforms the similarity scores into a condensed vector space that minimizes the distance of proteins from the same family. The similarity scores of a query to all profiles are used for its representation instead of considering only the top-ranking hit. In a protein family assignment task, FAMUS outperformed KEGG's native KofamScan for KEGG Orthology annotation and InterPro's InterProScan for PANTHER family annotation. We thus created four protein annotation models using protein families from the KEGG Orthology, InterPro family, OrthoDB, and EggNOG databases. All four models are available as a conda package and via our user-friendly web server, allowing users to annotate large-scale datasets. FAMUS is the first comprehensive and modular annotation framework based on contrastive learning. It supports both pre-defined and user-specific databases for tailored annotation, and can be easily integrated into any genomic and metagenomic analysis pipeline to facilitate accurate, large-scale functional annotation.
|
|
Scooped by
mhryu@live.com
March 10, 10:35 PM
|
Given that most microbes experience spatially structured environments, examining how such environments affect microbial growth and functions is paramount. Previous studies have shown that a spatially structured environment can impact microbial growth and interactions, and that microbial growth can create or magnify spatial structure. Here, we review some of these instances of past studies to develop a consistent framework that highlights the interplay between microbial interactions, spatial structure of the environment and spatial organization of microbes. We re-examine the level, degree and scale of spatial structure with regard to the phenomena and biological processes of interest. We then discuss how mathematical models can reveal the contribution of the spatial structure to community assembly and coexistence. Lastly, we offer an outlook on important steps for the progress of this field.
|