Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:10 PM
|
Transcriptional terminators are key to defining transcript boundaries, ensuring mRNA maturation, and maintaining expression stability, yet they remain underexplored in filamentous fungi. Here, we systematically benchmarked 15 heterologous 3′ terminators in Aspergillus oryzae by measuring protein output via mCherry fluorescence and transcriptional readthrough based on a quantitative readthrough index (RTI) derived from RT-qPCR. Across the tested terminators, mCherry expression varied more than 5-fold and RTI spanned over 50-fold, indicating substantial functional diversity. Expression strength and termination efficiency were only weakly correlated (Spearman ρ = −0.39, p = 0.165), indicating that these two properties are largely independent and can be optimized separately. Strong terminators -TactA, Txyn1, TmutA and TtrpC combined good mCherry expression (>ΔT) and small readthrough (RT < 0.15). In contrast, short synthetic sequences (≤70 bp) showed low mCherry output accompanied by transcriptional leakage. Interestingly, widely used toolkit elements such as Ttef1 and Tcyc1 performed poorly in A. oryzae, highlighting the importance of empirical characterization over assumed transferability. Polyadenylation site mapping by 3′ rapid amplification of cDNA ends (3′RACE) reveals high-performing terminators that possess focused cleavage sites, continuous poly(A) tails, and recognizable A[AT]TAAA-like motifs with moderately AU-rich upstream regions, whereas weak terminators exhibited dispersed cleavage and sparse upstream sequence elements. Targeted disruption of the canonical AATAAA hexamer in TactA did not significantly impair expression or termination efficiency, demonstrating that the canonical PAS is not strictly required when AU-rich upstream sequence elements are present. Terminator performance rankings were robust across three carbon sources and were confirmed using a secreted NanoLuc luciferase reporter, demonstrating that functional behavior is largely sequence intrinsic. These findings demonstrate a functional terminator architecture and suggest a practical guideline for terminator selection in A. oryzae.
|
|
Scooped by
mhryu@live.com
Today, 10:30 AM
|
Accurately predicting protein-protein interactions (PPIs) in dimeric complexes remains a fundamental challenge in computational biology. Although existing PPIs prediction models, such as AlphaFold-Multimer (AF-Multimer) and AlphaFold3 (AF3), have achieved impressive performance, they still suffer from unsatisfactory accuracy due to the limited availability of protein dimer structures, whose collection is both expensive and labor-intensive. Here, we introduce a simple yet effective pre-training method, termed split and merge proxy (SMP), that leverages abundant monomeric proteins to simulate various PPIs tasks for the first time. Specifically, SMP constructs pseudo-dimers by splitting monomer data into two subunits, referred to as pseudo-receptors and pseudo-ligands, and trains models to merge them back by predicting their pseudo interactions (e.g., contact or docking). This proxy task enables large-scale pre-training without additional cost. Models pre-trained with SMP and subsequently fine-tuned on real protein dimer datasets demonstrate consistently improved accuracy and generalization across multiple benchmarks, surpassing strong baselines. Notably, SMP delivers more accurate structure predictions than both AF-Multimer and AF3 on several CASP15 dimer targets. Our findings highlight SMP as a scalable strategy for harnessing monomeric data to advance protein complex modeling, providing insights into the linkage between monomers and multimers. Accurate prediction of protein-protein interactions is limited by the scarcity of high-quality complex structures. Here, authors introduce SMP, a strategy that leverages pseudo-dimers derived from monomers to improve accuracy and generalization across diverse protein interaction applications.
|
|
Scooped by
mhryu@live.com
May 31, 3:53 PM
|
Non‑alcoholic beers (NABs) are a rapidly expanding market. However, producing high‑quality products remains challenging because ethanol strongly shapes beer flavor and mouthfeel. Biological production methods, which restrict alcohol formation during fermentation, offer advantages over physical methods, which remove alcohol after fermentation. However, they are limited by the performance and diversity of available yeasts. Recent advances in bioprospecting, breeding, hybridization, adaptive evolution, and genetic engineering reveal how different yeast lineages and trait-development strategies can balance reduced ethanol production with desirable aromatic complexity. Yeasts with naturally limited sugar utilization and favorable aroma‑forming capacities are emerging as promising foundations for next‑generation NAB production. Progress in this field will depend on integrating biological strategies with standardized phenotyping, deeper exploration of natural diversity, and emerging computational tools for predicting flavor and guiding targeted strain improvement. Together, these developments outline a path toward rational, data‑driven design of superior yeasts tailored for NAB production.
|
|
Scooped by
mhryu@live.com
May 31, 9:50 AM
|
Conventional methods for monitoring toxic heavy metals typically require sophisticated laboratory instrumentation and leave a critical gap for rapid, on-site detection. Herein we present Transcription-factor-Occluded Nick Extension or TONE, a rapid, isothermal biosensing platform for heavy metal detection that exploits allosteric transcription factor (aTF) regulation to gate DNA strand-displacement amplification. In this approach, operator sequences modified with deoxyinosine (dI) substitutions are employed. When bound by an aTF, the dI sites are shielded from cleavage by Endonuclease V. Upon exposure to target heavy metals, the aTF dissociates, permitting the enzyme to nick the DNA and trigger a strand-displacement reaction. The amplified DNA is then detected via an instrument-free lateral flow assay, delivering a visual readout within 25 minutes at room temperature. We demonstrate the utility of TONE using the TetR aTF and further adapt the assay for copper and lead detection through the transcription factors CsoR and CadC, respectively, achieving detection limits as low as 40 nM and 80 nM, respectively. TONE offers a sensitive, low-cost, and field-deployable solution for environmental monitoring and other applications requiring rapid heavy metal analysis.
|
|
Scooped by
mhryu@live.com
May 31, 9:34 AM
|
This study presents a mathematical framework for investigating the dynamics of coexistence and competition among heterotrophic microbes across different time scales. Focusing on metabolic interactions, we examine how three strategies: public metabolizing, private metabolizing, and cheating, shape population behavior. The framework integrates generalized Lotka-Volterra dynamics with evolutionary game theory to capture the effects of resource exchange, particularly glucose made available by public metabolizers and sucrose as a shared substrate driving population growth. Game-theoretic payoffs encode ecological costs and benefits, enabling analysis of frequency-dependent interactions among strategies. To capture evolutionary realism, we implement laboratory-inspired simulations in which strategies can switch between generations, mimicking mutation or phenotypic plasticity in microbial populations. These eco-evolutionary dynamics reveal conditions under which all three strategies coexist at interior equilibria and show how variation in growth advantages and, illustratively, phenotype-switching perturbations produce evolutionary shifts. Numerical analysis identifies ecological thresholds and fitness asymmetries that determine system robustness, long-term coexistence, and the persistence of a synthetic, cross-kingdom system linked by nutrient exchange. Together, these insights provide general principles for microbial coexistence and offer design guidelines for ecosystem engineering, biotechnological applications, and the construction of stable synthetic communities under ecological and evolutionary constraints.
|
|
Scooped by
mhryu@live.com
May 31, 1:06 AM
|
The RNA polymerase that transcribes photosynthetic genes in the plant chloroplast is the largest known transcription enzyme across all domains of life, comprising 21 subunits of bacterial and eukaryotic origin. Recent structural analyses revealed that the core polymerase, inherited from the cyanobacterial ancestor of the chloroplast, is encased by subunits unlike known bacterial transcription proteins. This insight into the composite nature of the complex provides clues about how the polymerase interacts with transcription regulatory elements of both bacterial and eukaryotic origin. Here, we summarize insight from recent structural and biochemical data on chloroplast and cyanobacterial transcription complexes. We analyze the available evidence on the mechanisms of chloroplast Sigma factors, NusG, mTERF proteins, kinases and alarmones, considering prevailing models of transcription control in bacteria and mitochondria to produce new hypotheses on the molecular basis of chloroplast transcription control to be characterized in the future.
|
|
Scooped by
mhryu@live.com
May 31, 12:59 AM
|
Prokaryotes harbor a diverse spectrum of extrachromosomal elements (ECEs), which are intracellular replicons maintained independently of the primary chromosome. Historically, the ECE research field has focused on relatively small ECEs, such as plasmids. However, the advent of long-read sequencing has revealed that prokaryotes also harbor various types of giant ECEs, spanning hundreds of kilobases to over 1 Mb, that were not hitherto recognized. In this review, we describe how long-read sequencing has enabled the discovery of giant ECEs and compare the genetic architectures and functional repertoires of several recently characterized examples. The functions of most genes in these ECEs remain uncharacterized, and current computational tools frequently misclassify or overlook them. We further discuss how the discovery of these giant ECEs challenges existing classification frameworks that attempt to distinguish megaplasmids, chromids, and chromosomes. Together, these findings highlight giant ECEs as a largely unexplored layer of microbial genetics, whose characterization will have broad implications for our understanding of microbial adaptation and horizontal gene transfer.
|
|
Scooped by
mhryu@live.com
May 31, 12:37 AM
|
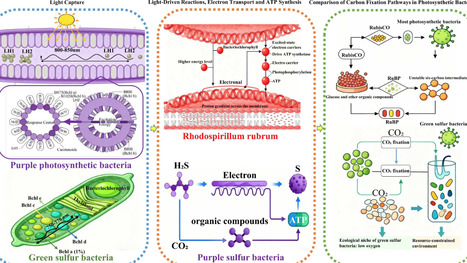
Photosynthetic bacteria, a diverse group of microorganisms endowed with distinctive photosynthetic mechanisms, have attracted increasing attention on account of their substantial ecological functions and extensive application prospects. This review comprehensively summarizes the classification, physiological and biochemical characteristics of photosynthetic bacteria, and explores their pivotal functions in carbon, nitrogen, and sulfur cycling. Moreover, the potential applications of photosynthetic bacteria in biotechnology, including wastewater treatment, agriculture, aquaculture, and bioenergy production, are systematically presented. The latest research methods, current research trends, and future development directions are also analyzed in detail. Despite their considerable potential, challenges like high-cost large-scale cultivation and strain instability still exist. A more comprehensive understanding of photosynthetic bacteria will contribute to their more effective utilization in addressing environmental and energy problems.
|
|
Scooped by
mhryu@live.com
May 31, 12:29 AM
|
Nitrogen acquisition is pivotal for plant growth. In soil ecosystems, bacterial interactions promote nitrogen assimilation and rhizosphere colonization. However, the mechanisms underlying the interactions between nitrogen-fixing microorganisms and their neighboring organisms in the environment remain unclear. Here, we demonstrate that Bacillus velezensis BRI3, a rhizosphere-derived strain, forms microbial synergy with Stutzerimonas stutzeri A1501, functioning as a facilitator. This microbial synergy greatly increases the nitrogen-fixation by 3.2-fold and rhizosphere colonization capabilities by 2.3-fold of A1501, collectively promoting plant growth in the rhizosphere. In this study, for the first time, we propose that surfactin produced by BRI3 regulates interactions among this bacterial consortium by stimulating A1501 biofilm formation. This discovery enhances the understanding of metabolic interactions between nitrogen-fixing bacteria and their neighboring organisms. Overall, we propose a strain interaction paradigm that offers a novel framework for improving nitrogen utilization and crop yield.
|
|
Scooped by
mhryu@live.com
May 31, 12:21 AM
|
Next-generation sequencing has revolutionized microbiome research, yet the transition from taxonomic to functional profiling remains a major technical challenge. While marker gene sequencing provides a widely accessible ecological view, it often lacks the resolution for actionable insights. This perspective argues that shifting to whole metagenomic sequencing is essential for mapping functional potential, such as antimicrobial resistance, and metabolic pathways. However, we identify a critical bottleneck: excessive multiplexing. High multiplexing ratios reduce the number of unique molecules per sample, leading to high duplication rates and the stochastic dropout of low-abundance genes. We demonstrate that functional profiles are far more sensitive to these library complexity issues than taxonomic ones. We recommend prioritizing total sequencing depth and reducing multiplexing to ensure sufficient unique coverage. Additionally, adopting long-read or hybrid architectures is vital for providing the genomic context necessary for strain-level resolution. These optimizations are prerequisites for robust global microbiome synthesis and translational science.
|
|
Scooped by
mhryu@live.com
May 31, 12:08 AM
|
Two deeply conserved protein families, Argonaute (Ago) and Schlafen (SLFN), play defense roles in diverse prokaryotes and eukaryotes, including humans. Here, we identify a monophyletic group of proteins broadly distributed across bacteria and archaea that fuse a SLFN domain with an Ago core and a GHKL-family ATPase. Using SLFN-pAgo from Runella zeae as a model, we show that these proteins protect bacterial cells against bacteriophages by employing the Ago core as a guide-dependent sensor and the SLFN domain as a nuclease effector to induce abortive infection via tRNAs cleavage. Structural and biochemical analyses reveal that ATP binding by the GHKL domain drives tetramerization of SLFN-pAgo, reconstituting the canonical SLFN nuclease architecture found in mammalian proteins. Prior to target detection, non-canonical guide RNA binding induces the formation of long helical filaments, locking the SLFN domains in an inactive configuration. Guide-dependent recognition of complementary target DNA triggers massive structural rearrangements leading to filament disassembly and the induction of SLFN nuclease activity. Together, our findings uncover a new antiphage system that employs reversible guide- and ATP-mediated oligomerization to strictly regulate cooperation between an Ago sensor and a SLFN RNAse effector.
|
|
Scooped by
mhryu@live.com
May 30, 11:59 PM
|
Chloromethane, a toxic gas primarily produced naturally, contributes to stratospheric ozone destruction. The anaerobic acetogen Acetobacterium dehalogenans can utilize chloromethane as a carbon and energy source, but the associated dehalogenase/methyltransferase has remained elusive. Through comparative transcriptomics we identify a gene cluster, cdmBCA, which encodes a corrinoid-dependent methyltransferase system distinct from the characterised Cmu system used for chloromethane degradation in aerobic methylotrophs. Biochemical characterization reveals that the Cdm system reacts with other haloalkanes, but not with methoxylated aromatics, unlike closely related O-demethylases. X-ray structural analysis of the protein CdmB shows a hydrophobic channelling system directing haloalkanes towards cobalamin-dependent activation. Homologous proteins are found in anaerobic prokaryotes, particularly within the phyla Bacillota and Asgardarchaeota, suggesting previously unidentified microbial transformation of chloromethane in the environment. Discovery of the Cdm dehalogenation/methyltransferase system sheds light on the microbial contribution to the global chloromethane cycle. Chloromethane, a toxic gas primarily produced naturally, contributes to ozone destruction. Here, the authors identify and characterise an enzyme system that dehalogenates chloromethane and appears to be specific to anaerobic microorganisms.
|
|
Scooped by
mhryu@live.com
May 30, 11:03 PM
|
Iron-sulfur clusters are versatile protein cofactors involved in diverse biological processes, but their role in hydrogen sulfide/hydrosulfide (H2S/HS-) sensing remains largely unexplored. Here, we report that the Bacillus licheniformis sensor kinase NreB contains an unusual [2Fe-2S] cluster within its PAS domain. A 1.52-Å crystal structure reveals a distinct coordination geometry where three conserved cysteine residues and a non-cysteinyl sulfur ligand stabilize the cluster. Biochemical and native mass spectrometry support assignment of the predominant ligand state as -SH and show enrichment of -SSH/SSOH-like state upon sulfide exposure in the presence of O2, correlating with increased NreB kinase activity. Electron paramagnetic resonance spectroscopy shows that the cluster retains its oxidized [2Fe-2S]2+ state during sulfide-sensing. Molecular dynamics simulations further reveal transient solvent and HS- accessibility to the buried cluster, providing a physical basis for ligand entry. Here, we show that bacteria sense sulfide via a three-cysteine-coordinated Fe-S cluster with a labile sulfur ligand. Sulfide is an important bacterial signal, but how bacteria detect it remains unclear. Here the authors show that NreB uses an unusual iron-sulfur cluster with a labile sulfur ligand to sense sulfide and regulate kinase activity.
|
|
|
Scooped by
mhryu@live.com
Today, 10:59 AM
|
Engineered therapeutic microbes for intestinal inflammation must be capable of gut colonization, sensitive detection of disease-associated biomarkers and targeted delivery of therapeutic molecules. Examples of microbes displaying all three characteristics are limited. Here we engineered Bacteroides thetaiotaomicron, a human gut commensal bacterium with colonization ability, colonic tropism and innate anti-inflammatory properties, as a chassis to create programmable bacterial strains termed Btbots. We developed genetic circuits that were integrated into the bacterial chromosome to sense two intestinal inflammation biomarkers, deoxycholic acid (DCA) and nitric oxide (NO), and to enhance surface display and secretion of therapeutic molecules in response. Btbots with these biosensors were developed that either display trefoil factor-3 to facilitate mucosal repair or secrete interleukin-35 to suppress inflammation. These Btbots sensed DCA and NO biomarkers and released therapeutic agents, alleviating colitis and modulating the gut microenvironment and microbiota in mouse models. This work establishes a proof of concept for localized sensing and consequent therapeutic molecule release for gastrointestinal applications, whose clinical potential awaits further investigation. Engineered Bacteroides thetaiotaomicron, Btbots, use a genetic AND circuit to sense nitric oxide and deoxycholic acid as inflammatory signal in the gut to release therapeutic molecules, TFF3 and IL-35, that alleviate colitis in mice.
|
|
Scooped by
mhryu@live.com
Today, 10:22 AM
|
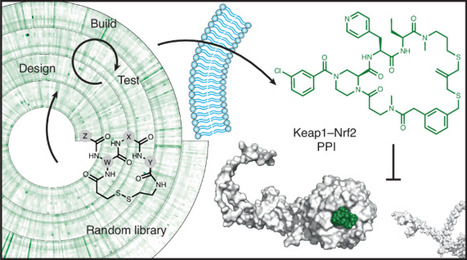
Small, nonpolar cyclic peptides can both bind challenging targets and cross cell membranes, making them attractive for addressing currently undruggable targets such as many protein–protein interactions (PPIs). However, developing such compounds de novo without prior information about lead structures such as natural ligands or fragments remains a notable challenge. Here we show that functional screening of structurally highly diverse cyclic peptide libraries synthesized at nanomole scale allows identification of sub-kDa inhibitors of a PPI. By screening 15,360 fully random cyclic peptides, we were able to identify an inhibitor of the E3 ligase adaptor Keap1 and its substrate Nrf2. Optimization by rapid design–build–test cycles produced a membrane-permeable compound active in live cells. This study demonstrates that large, diverse cyclic peptide libraries can enable the discovery of cell-permeable PPI inhibitors from the ground up, providing a way to harness the powerful modality of small cyclic peptides to address often difficult-to-target intracellular interactions. Membrane-permeable cyclic peptides offer access to difficult intracellular targets but discovery remains challenging. Here the authors show that cell-active cyclic peptides can be identified by screening sufficiently large and diverse libraries of small synthetic peptides.
|
|
Scooped by
mhryu@live.com
May 31, 9:57 AM
|
Bacteria are able to coordinate cell growth and genome replication in different growth conditions.The DNA-binding protein DnaA is responsible for determining initiation of replication, thereby playing a central role in this coordination. Theoretical and experimental studies have shown that stability of the cell cycle requires an ultrasensitive response, i.e., a sharp dependence of the initiation firing rate on the cell volume. However, the source of such ultrasensitivity remains elusive. In this work, we elucidate how the structure and binding affinities of the DnaA regulatory system determine its ultrasensitive response. Our theory sets precise constraints on inding parameters, that are necessary for cell cycle stability. Our findings show how the variety of regulatory mechanisms of the DnaA system are required for ultrasensitivity across growing conditions.
|
|
Scooped by
mhryu@live.com
May 31, 9:40 AM
|
We evaluate whether tryptophan (W), widely thought to be the last of the 20 canonical amino acids added to the genetic code, was already present in the Last Universal Common Ancestor (LUCA). We reconstruct the evolutionary history of tryptophanyl-tRNA synthetase (WRS), the enzyme that attaches W to its tRNA, and the related tyrosyl-tRNA synthetase (YRS). We identify and exclude sequences derived from ancient recombination between archaeal and bacterial YRSs. Diverse rooting methods, including a novel approach exploiting time non-reversible evolution, all place the root between bacterial and archaeal YRS rather than between YRS and WRS. This supports post-LUCA WRS origination in Archaea, followed by its horizontal transfer to Bacteria. However, ancestral sequence reconstruction suggests that Archaea were depleted for W while Bacteria were not, and enzymes essential for W biosynthesis emerged in Bacteria. This suggests that W usage originated in Bacteria, with later WRS emergence in Archaea allowing the archaeal genetic code to converge with the bacterial code. The universality of the genetic code is usually attributed to common descent from LUCA, but the final step making the code universal was instead achieved by horizontal gene transfer. This gives credence to similar mechanisms for earlier steps in genetic code evolution.
|
|
Scooped by
mhryu@live.com
May 31, 1:08 AM
|
As mobile genetic elements, transposons play a crucial role in the adaptive evolution and genome engineering of industrial microorganisms. Their applications range from high-throughput functional genomics, enabling systematic genotype-phenotype mapping via transposon sequencing, to the construction of random integration libraries for chassis development and directed evolution. Despite the emergence of precise editing tools such as CRISPR-Cas, transposon technology remains indispensable in non-model industrial strains owing to its operational simplicity and high efficiency. Recent discoveries of novel transposon systems, along with their functional enhancement, have further expanded their utility. Looking ahead, integrating transposon technology with strategies such as AI-assisted design and CRISPR-Cas-based systems will greatly advance our ability to decipher and engineer industrial microbial cell factories. This review summarizes the principles, challenges, and opportunities of transposon-associated technologies in industrial microorganisms, offering new insights into their roles in industrial biotechnology.
|
|
Scooped by
mhryu@live.com
May 31, 1:04 AM
|
Our current industrial, agricultural, and medical practices exploit the extraordinary biodiversity generated through billions of years of natural evolution. Despite their high fitness in native habitats, biomolecules and organisms are often not optimally suited for industrial and medical use. Synthetic evolution leverages technologies such as DNA synthesis, CRISPR–Cas engineering, and synthetic biology to enable continuous in vivo mutagenesis of biomolecules or organisms to improve specific desirable characteristics. This review presents the latest mutagenesis toolkits classified by mutational scale: genome-wide, medium-scale, and site-specific, each tailored to different application scenarios. We discuss the mechanisms and capabilities underlying each scale, analyze current limitations, and highlight the untapped potential of next-generation gene-editing technologies, high-throughput screening, and artificial intelligence in advancing synthetic evolution.
|
|
Scooped by
mhryu@live.com
May 31, 12:42 AM
|
Programmable control of microbial gene expression by plant hosts could enable a new generation of precision agricultural biotechnology. Here, using O-methyl-L-tyrosine (OMY) as a model compound, we establish non-standard amino acids (nsAA) as a platform for plant-based control of associated microbial activity. We use genetic code expansion to engineer OMY-dependent control of protein synthesis in the soil bacterium Bacillus subtilis. Then, we engineer agronomically diverse plants, including Arabidopsis, tomato and poplar, to biosynthesize OMY. We show that plant-derived OMY can stimulate gene expression in both model and wild soil bacteria and demonstrate how inducible and tissue-specific expression of a single biosynthetic enzyme by the plant enables tight, on-demand control over microbial activity. This work establishes nsAAs as a tool for programming plant-microbe partnerships.
|
|
Scooped by
mhryu@live.com
May 31, 12:35 AM
|
Cyclic di-GMP (c-di-GMP) is a key bacterial second messenger that regulates a wide range of cellular processes, including biofilm formation and virulence. Multi-domain one-component systems regulate c-di-GMP synthesis and turnover in response to external signals. Periplasmic sensing of environmental cues is performed by versatile but conserved sensory domains, including members of the CHASE4 superfamily. Here, we explore the promiscuity of the CHASE4 domain in c-di-GMP signal transduction in Pseudomonas aeruginosa by analyzing two CHASE4-containing transducers from the virulent PA14 strain, namely PA14_53310 and PA14_37690. With the integration of biochemical and biophysical methods, such as UV-Vis spectroscopy, circular dichroism, and isothermal titration calorimetry, we demonstrate that the CHASE4 domains of these proteins possess different ligand specificities. We show that PA14_53310 is a diguanylate cyclase, and that its enzyme activity is controlled by heme binding to its periplasmic CHASE4 domain. The PA14_37690 CHASE4 domain, on the other hand, does not bind heme, but likely recognizes copper, indicating a role in metal ion sensing. A comparison with the PAO1 counterpart, that is, PA0847 and PA2072, is discussed and the divergences highlighted. This work demonstrates that the CHASE4 domain is a multifunctional sensory module that can be calibrated to detect a range of environmental cues, thus providing a mechanism for c-di-GMP signaling refinement in P. aeruginosa. These results shed light on the molecular basis for the functional diversification of CHASE4-containing one-component systems and their role in bacterial adaptation.
|
|
Scooped by
mhryu@live.com
May 31, 12:24 AM
|
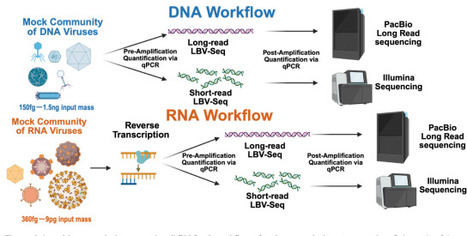
Genome-resolved virome analysis remains inaccessible for many samples, including those with clinical relevance, because viral nucleic acid recovered after enrichment is often too scarce to support de novo genome assembly. As a result, many analyses are limited to sparse read-level detection, which cannot recover divergent viruses, resolve strains, or interpret gene-level variation. Here, we developed Low Biomass Viral Sequencing (LBV-Seq), a workflow that couples low-input viral sample handling with modified primary template-directed amplification and short- or long-read sequencing to enable de novo reconstruction of DNA and RNA viral genomes from sub-femtogram to nanogram inputs. LBV-Seq reproducibly captures the same relative community composition, amplifies diverse viruses, and achieved broad genome coverage across nearly all targets regardless of viral genome structure, Baltimore class, abundance, or input mass. Short-read assemblies recovered near-complete genomes from femtogram-scale inputs. Long-read sequencing provided orthogonal support for genome structure, with PacBio HiFi reads spanning large portions of viral genomes and, in some cases, complete small viral genomes. Applied to virus-enriched human duodenal biopsy eluates, LBV-Seq provided proof-of-feasibility for recovering both bacteriophage and eukaryotic viral genomes from low-input biopsy-derived material. In the eluates tested, LBV-Seq recovered co-occurring Alphatorquevirus and Betatorquevirus genomes estimated to be present at roughly 10 copies/μL and at viral masses below 0.1 fg/μL. LBV-Seq enables genome-resolved virome analysis in samples previously limited to detection-based viromics, supporting viral discovery and strain-resolved analyses in settings where viral mass is low, including viruses enriched from human tissue biopsies.
|
|
Scooped by
mhryu@live.com
May 31, 12:16 AM
|
5-Methylcytosine (5mC) plays an important role in gene regulation and development. Although nanopore sequencing has enabled direct detection of 5mC, existing methods still face several limitations, including poor generalization across species and sequence contexts (CpG/CHG/CHH), as well as suboptimal integration of sequence and current signals. Here, we present MethyNano, a deep learning framework incorporating a contrastive learning strategy to detect 5mC from nanopore reads. By encouraging more discriminative and stable representations, the contrastive objective improves the model’s sensitivity to rare sequence contexts and reduces its prediction uncertainty in challenging regions. Across datasets from A. thaliana, O. sativa, and H. sapiens, our model achieves superior performance on key metrics compared with other existing methods. Extensive cross-species and cross-motif experiments demonstrate the robust generalization performance of MethyNano, while dimensionality-reduction visualizations of learned features provide an intuitive view of the model’s efficient representation capability. Moreover, our ablation studies show that MethyNano’s architecture enables more effective integration of critical features, leading to higher predictive accuracy.
|
|
Scooped by
mhryu@live.com
May 31, 12:06 AM
|
Tumor immunotherapy has revolutionized cancer treatment by harnessing the immune system to recognize and eliminate malignant cells. However, limited response rates, systemic toxicity, and tumor heterogeneity remain major challenges. Synthetic gene circuits — engineered genetic programs capable of sensing, integrating, and responding to biological cues — offer a powerful strategy for developing next-generation ‘smart’ therapeutics. In this review, we summarize recent advances in synthetic gene circuits that enable smart and controllable tumor immunotherapy. We then discuss the design principles of these circuits, including small-molecule-inducible, physically responsive, and endogenous signal-responsive systems. Representative applications in tumor immunotherapy are highlighted, including programmable immune cells, microbial platforms, and tumor-intrinsic circuits that enable context-dependent activation, localized cytokine or effector release, and tumor-selective immunogenic cell death. Collectively, these advances illustrate the potential of synthetic gene circuits to improve specificity, safety, and therapeutic efficacy, paving the way for precision cancer immunotherapy and the next generation of cancer therapeutics.
|
|
Scooped by
mhryu@live.com
May 30, 11:40 PM
|
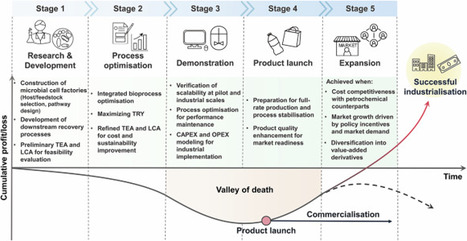
Biomanufacturing offers a sustainable alternative to petrochemical manufacturing by harnessing engineered microorganisms to convert renewable feedstocks into diverse value-added products. Systems metabolic engineering has driven its advancement, enabling rational design of microbial strains with enhanced performance and robustness. Despite these achievements, large-scale commercialization remains limited, highlighting the persistent gap between laboratory success and industrial deployment. We examine the challenges and opportunities in advancing biomanufacturing towards industrial implementation through two cases, succinic acid and polyhydroxyalkanoates, that exemplify products nearing, yet not fully reaching, commercial maturity. Finally, we discuss how integrating artificial intelligence with systems metabolic engineering can accelerate sustainable biomanufacturing. Biomanufacturing holds promise as an alternative to the petrochemical industry, with benefits including increased sustainability as well as improved supply chain resilience, a factor of particular importance in times of geopolitical instability. Here the authors explore both the challenges and opportunities in progressing biomanufacturing from laboratory successes towards industrial-scale deployment.
|
















![A labile sulfur ligand in a three-cysteine-coordinated [2Fe−2S] cluster mediates sulfide sensing in NreB | Ncm | RMH | Scoop.it](https://img.scoop.it/dkvy_QUIqNNnnmEW_-IlPzl72eJkfbmt4t8yenImKBVvK0kTmF0xjctABnaLJIm9)