Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:52 PM
|
RNA interference (RNAi) has emerged as a promising approach to sustainable crop protection. Extensive proof-of-concept studies have led to the approval of the first sprayable plant protection products in the United States and China, with Europe currently evaluating them. Although gene silencing mechanisms are among the most extensively studied processes in molecular biology, with two Nobel Prizes recognizing their discovery, the optimization of delivery systems and field performance remains an area currently undergoing extensive development. The uptake, stability, and efficacy of double-stranded RNA (dsRNA) are influenced by species-specific and environmental factors, introducing variability that must be understood in order to select robust targets, design effective dsRNA, and assess risk. Although these knowledge gaps remain, they are increasingly addressed through systematic experimental and technological advances. This review summarizes the current knowledge on RNAi mechanisms in plants, fungi, and insects, emphasizing the differences in dsRNA uptake and processing between species. We highlight advances in formulations and delivery technologies, discuss how regulatory and ecological questions are being systematically investigated, and present examples of approved products that demonstrate the approach's feasibility and safety. Finally, we outline how remaining uncertainties can be addressed through targeted research and risk-mitigation strategies and how RNAi technologies can be incorporated into comprehensive pest and disease management systems.
|
|
Scooped by
mhryu@live.com
Today, 12:32 PM
|
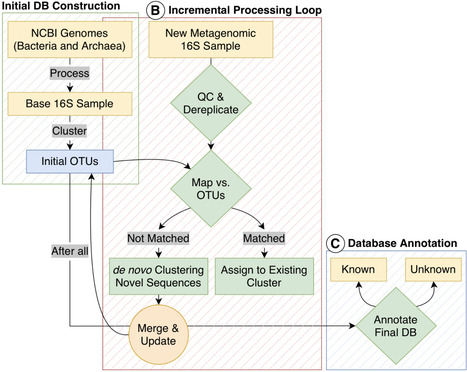
The exponential growth of public metagenomic datasets offers an unprecedented opportunity to explore microbial diversity. However, analyzing this vast amount of data presents significant computational challenges. While shotgun metagenomics provides deep functional and taxonomic resolution, its high cost still limits its application. On the other hand, 16S rRNA gene sequencing remains a cost-effective and widely used alternative, but tools are needed to maximize its discovery potential. Traditional clustering is not scalable, obstructing the creation of a comprehensive and continuously updated catalog of microbial life from 16S data. We developed a reproducible and scalable Snakemake pipeline for the incremental clustering of 16S rRNA amplicons. The workflow begins by constructing a reference database from bacterial and archaeal genomes. It then processes 16S rRNA samples sequentially. For each new sample, sequences are first mapped against the existing cluster centroids. Sequences that match known centroids are assigned accordingly, while unmapped sequences are clustered independently to form novel operational taxonomic units (OTUs). These new centroids are then merged with the existing database, allowing it to grow dynamically without the need for computationally prohibitive all-at-once re-clustering. Our pipeline enables the efficient and continuous expansion of a 16S rRNA cluster database. By processing a large corpus of public 16S rRNA samples, we generated a comprehensive atlas of tens of thousands of OTUs. A significant fraction of these clusters, particularly at the genus and family levels, were classified as unknown. This work provides a powerful, open-source tool for large-scale analysis of 16S rRNA samples. The incremental clustering strategy overcomes the scalability limitations of traditional methods, allowing researchers to leverage public data and discover novel microbes in their own microbiome samples.
|
|
Scooped by
mhryu@live.com
Today, 12:19 PM
|
Sponge RNAs (spRNAs) play an important regulatory role in bacterial small RNA (sRNA) networks, but their engineering and quantitative systems-level properties are unexplored. Here, we design, build, and quantitatively characterize synthetic spRNA-based gene circuits in E. coli. We establish multiple design strategies for synthetic spRNAs, engineering the first synthetic spRNAs. We show that these synthetic spRNAs can reversibly de-repress sRNA-regulated gene expression, demonstrate tuneable control of gene expression, and extend these designs to multi-target regulation. Through the use of time-resolved continuous-culture characterization in Chi.Bio together with absolute fluorescent protein quantification, we generated a quantitative dynamical dataset for model fitting and mechanistic analysis. Sequential model development showed that recapitulating the observed circuit dynamics required incorporation of Hfq-mediated resource competition, often overlooked in models of sRNA-based synthetic gene circuits. The extended model captured promoter, sRNA, and sponge circuit behavior and was used to investigate quantitative properties of spRNA-mediated regulation, the first such quantitative investigation of spRNA-based regulation. Model-based quantitative investigations further suggest that spRNAs can tune response functions, modulate thresholds and leakiness, alter response times, improve disturbance rejection in some regimes, increase effective specificity, and buffer regulatory output against sRNA mutation. Together, these results establish synthetic spRNAs as a new post-transcriptional tool for bacterial synthetic biology and provide a quantitative framework for understanding natural and engineered spRNA-mediated regulation.
|
|
Scooped by
mhryu@live.com
Today, 12:12 PM
|
Environmental contamination by heavy metals, organic pollutants, and industrial waste poses a pressing global challenge that demands advanced, sustainable remediation technologies. Conventional remediation approaches, including soil excavation and chemical treatment, are costly, generate secondary pollutants, and lack versatility across contamination types. Phytoremediation offers a cost-effective and environmentally safe alternative; however, it is constrained by slow pollutant uptake rates, limited degradation efficiency for persistent organic compounds, and inadequate real-time monitoring capabilities. This review presents a novel interdisciplinary framework that synergistically integrates phytoremediation with nanotechnology, biotechnology, and information technology (IT) to overcome these inherent limitations. Nanotechnology facilitates pollutant detoxification through engineered nanoparticles (NPs) that enhance metal bioavailability and degrade organic pollutants. Biotechnology encompassing genetic engineering and microbial-assisted remediation enhances plant metal accumulation, stress tolerance, and pollutant degradation pathways. IT approaches, including artificial intelligence (AI), remote sensing, and big data analytics, enable real-time monitoring, adaptive strategy optimization, and predictive modelling of remediation outcomes. Together, these technologies constitute a multi-dimensional remediation framework aimed at minimizing environmental damage and promoting ecological sustainability. This review synthesizes recent advancements, highlights key knowledge gaps, and identifies future research directions for next-generation phytoremediation.
|
|
Scooped by
mhryu@live.com
Today, 9:29 AM
|
lthough protein engineering and laboratory evolution have been used to optimize prime editors, we show that previous changes that improve prime editor efficiency also compromise protein stability and expression level, limiting performance. To address these limitations, we apply structure-informed artificial intelligence-guided methods such as the inverse-folding network ProteinMPNN to redesign the reverse transcriptase (RT)domains of engineered and evolved prime editors while preserving regions essential for catalysis. Redesigned RTs are extensively mutated, with 30–163 amino acid substitutions, and exhibit enhanced folding stability and soluble expression and up to twofold higher intracellular prime editor protein levels following mRNA delivery. Redesigned PE8 prime editors demonstrate enhanced editing efficiencies across multiple ex vivo contexts, including in several human primary cell types and via several delivery modalities. In mice, editing efficiency is up to 2.9-fold higher than that of state-of-the-art PE6, PE7 and PEmax prime editors. These findings demonstrate a generalizable approach for augmenting laboratory evolution to improve genome editing agents. A computational redesign strategy improves evolved prime editors.
|
|
Scooped by
mhryu@live.com
Today, 12:13 AM
|
Quantitative analysis of bacterial dynamics in time-lapse microscopy requires robust tracking pipelines, yet selecting and optimizing algorithms for specific experiments remains challenging. Indeed, microbiologists are confronted with numerous algorithms that must be carefully chosen and parameterized to achieve optimal tracking for their experiments. We present an automated methodology to determine optimal tracking configurations for microbiological applications. It is based on TrackMate 8, a novel version of the TrackMate Fiji plugin extended with microbiology-specific tools. Our approach systematically evaluates algorithm-parameter combinations optimizing biologically relevant metrics (e.g., cell-cycle accuracy, bacterial morphology) and includes: (1) integration of deep-learning algorithms (Omnipose, YOLO, Trackastra) adequate for bacterial images in TrackMate; (2) a TrackMate-Helper extension for parameter optimization; and (3) a tracking and segmentation editor for tracking ground-truth generation. We demonstrate the effectiveness of the methodology on two use cases showing its adaptability to diverse experimental conditions. This methodology enables microbiologists with a widely applicable, automated framework to optimize tracking pipelines, facilitating quantitative analysis in bacterial imaging.
|
|
Scooped by
mhryu@live.com
Today, 12:01 AM
|
Plants actively reshape the soil environment through their roots and associated microbes, creating lasting changes known as soil legacies that influence future plant generations via plant–soil feedbacks. While biotic factors such as pathogens and mutualists have received much attention, the chemical legacies, including water-soluble and volatile organic compounds, remain underexplored. These metabolites, produced by plants and soil microbes, modulate microbial communities, nutrient dynamics, and plant defenses, driving positive or negative feedbacks. This opinion article synthesizes recent evidence on soil chemical diversity, their role in legacy formation and persistence, while highlighting analytical challenges and promising applications in agriculture and ecology.
|
|
Scooped by
mhryu@live.com
May 20, 5:13 PM
|
The artificial intelligence (AI)-driven generation of genetic sequences holds transformative potential for addressing global challenges in agriculture, medicine, and bioenergy. Traditional approaches including hybridization, mutagenesis, and CRISPR-based editing enable targeted modification of endogenous DNA, yet remain constrained by natural sequence diversity. We here introduce PlantGFM, an application of the Hyena operator within a plant-oriented genomic foundation model, which was pre-trained on 10.84 billion nucleotides from 12 plant species and supports long-context (64 kb) prediction and sequence generation within a unified architecture. After fine-tuning on 10 annotated plant genomes, PlantGFM matched or exceeded the performance of specialized gene prediction tools. Beyond reproducing natural genes, it enables de novo design of novel candidates through the emergence capability of AI. Seven candidates selected through an AI–Human Knowledge fusion screening pipeline all showed transcriptional activity in Nicotiana benthamiana, two with stable protein expression—representing the first demonstration of DNA–RNA–protein expression of Large Language Model-generated sequences in plants. As a proof of concept, PlantGFM also exhibits emergent abilities in generating plant NLR genes. Our findings establish the feasibility of LLM technology for de novo plant gene design, providing a foundation for plant synthetic biology and AI-assisted breeding.
|
|
Scooped by
mhryu@live.com
May 20, 5:08 PM
|
Immobilised Metal Affinity Chromatography (IMAC) is widely used to purify his-tagged recombinant proteins from E. coli. However, endogenous contaminants with histidine clusters, such as GFAT and PDH E1 proteins, are often co-purified with the target protein. The low background strain LOBSTR-RIL has been previously engineered with mutated forms of SlyD and ArnA that exhibit reduced binding to Ni2+ resin. In this study, the LOBSTR-RIL strain was further modified to produce IMACulate(DE3), where we altered the glmS (encoding GFAT protein) and aceE (encoding PDH E1 protein) genes to reduce surface histidines. Proteins purified from this strain show reduced levels of GFAT contamination. No statistically significant difference was observed in the abundance levels of PDH E1 protein in the BL21(DE3)-RIL, LOBSTR-RIL and IMACulate(DE3) strains. The use of IMACulate(DE3) increases the purity of recombinant his-tagged protein preparations with no additional effort or expense.
|
|
Scooped by
mhryu@live.com
May 20, 5:02 PM
|
Pseudomonas aeruginosa and Klebsiella pneumoniae are gram-negative opportunistic pathogens that frequently colonize the human body and are major causes of infection. These bacteria are often co-isolated in polymicrobial urinary tract and lung infections, the latter of which is associated with increased disease severity and worse clinical outcomes. Despite their overlapping niches and clinical relevance, little is known about how these two pathogens interact and how those interactions influence human health. Given the growing recognition that microbial interactions are key drivers of disease, we investigated how P. aeruginosa and K. pneumoniae influence one another. We discovered an antagonistic interaction in which P. aeruginosa restricts the growth of K. pneumoniae. This inhibition is driven by phenazine production in P. aeruginosa, specifically the secondary metabolites pyocyanin and pyorubin, which are both necessary and sufficient to suppress K. pneumoniae growth. Using a diverse set of clinical isolates, we found that this antagonism is strain-dependent. Both the susceptibility of K. pneumoniae to phenazines and the ability of P. aeruginosa to restrict K. pneumoniae growth varies between strains. Moreover, the necessity of phenazine production is specific to the site of infection. Together, these findings demonstrate that strain background and environmental context are critical determinants of pathogen interactions. These findings reveal that both strain background and environmental redox conditions govern the ecological rules of pathogen interaction, providing a framework for predicting outcomes.
|
|
Scooped by
mhryu@live.com
May 20, 4:53 PM
|
Drug-resistant bacterial infections increasingly evade available antimicrobials, and many existing antimicrobial peptides remain limited by instability, toxicity to mammalian membranes and high manufacturing cost. Here we introduce a modular peptide technology that self-assembles into nanofibres on bacterial surfaces through a membrane-anchoring biphenyl group, a diphenylalanine linker and a cationic minimalistic peptide that together enable selective disruption of drug-resistant pathogens. Using cryogenic electron microscopy, molecular dynamics simulations, lipid-nanoparticle membrane mimetics and binding thermodynamics, we show that the peptide first forms short nanofibres that dock onto phosphatidylglycerol and then elongates into nanofibres that penetrate and destabilize the bacterial membrane without inducing resistance. The nanofibres retain antibacterial activity when recycled from killed bacteria and outperform vancomycin and several classical antimicrobial peptides against dense bacterial populations in vitro. In a mouse model of methicillin-resistant Staphylococcus aureus pneumonia, inhaled peptide nanofibres eradicate pulmonary infection and restore lung architecture without detectable toxicity. This modular strategy enables the design of potent, selective and low-cost antimicrobials. A modular peptide technology that self-assembles into nanofibres on bacterial surfaces through a membrane-anchoring biphenyl group, a diphenylalanine linker and a cationic peptide enables disruption of drug-resistant pathogens.
|
|
Scooped by
mhryu@live.com
May 20, 4:42 PM
|
Plants and animals respond to pathogens through pattern recognition receptor and Nod-like receptor proteins. Pathogens commonly use protein effectors to suppress host immunity for successful infection. However, the existence of non-protein effector classes remains comparatively understudied. Here we report an RNA–RNA recognition mechanism governing pathogen–host interaction, mediated by a regulatory RNA-encoding DNA sequence that separately generates two complementary regulatory RNAs. Specifically, a long non-coding RNA transcribed from this DNA region in the fungal pathogen Magnaporthe oryzae translocates into host rice cells and sequesters a complementary microRNA (miRNA), derived from a distinct host DNA region, thereby subverting host immunity. In turn, this rice-derived miRNA promotes disease resistance by repressing the expression of PKR1, a gene that encodes a negative regulator of host immunity. Sequestration of the host miRNA by the fungal long non-coding RNA releases PKR1 expression to facilitate fungal infection. We discovered that this regulatory RNA-encoding DNA sequence is probably widely present across diverse life species, mediating interactions between pathogens and their plant hosts. Collectively, our findings provide an approach for effective disease control using miRNAs derived from this important DNA region. A fungal long non-coding RNA from Magnaporthe oryzae translocates into rice cells to sequester a host microRNA that normally represses PKR1, a negative immunity regulator, thereby facilitating infection and revealing a widespread RNA-based pathogen–host interaction mechanism.
|
|
Scooped by
mhryu@live.com
May 20, 4:27 PM
|
Temperate bacteriophages are ubiquitous viruses that co-evolve with their bacterial hosts. They are defined by their ability to undergo two distinct life cycles: the lytic cycle, in which the phage produces more viral copies and kills the host, and the lysogenic cycle, in which the temperate phage exists as a prophage in the host. Temperate phages have long served as fundamental models in microbiology, genetics and evolutionary biology research, and their life cycles are among the most thoroughly characterized in virology. Historically, the phage life cycle was viewed primarily through the lens of how excision, replication and packaging drive the formation of infective particles. Although it captures the central processes of the phage life cycle, this narrow perspective overlooks the full range of interactions with the host and with other mobile genetic elements. In this Review, we re-examine the temperate phage life cycle in light of emerging insights that expand on this framework with unanticipated complexities. We argue that many properties of (pro)phages should be viewed as integral parts of their life cycle instead of being discussed separately. This holistic view is important to fully appreciate the intricacies of the temperate phage life cycle and the key roles of these viruses in microbiology and biotechnological applications. The life cycle of temperate bacteriophages involves lytic or lysogenic cycles and has historically served as a model for studying genetic regulation. This Review provides an updated overview of these cycles and highlights their complexities, supporting a greater appreciation of the ecological roles of temperate phages.
|
|
|
Scooped by
mhryu@live.com
Today, 12:36 PM
|
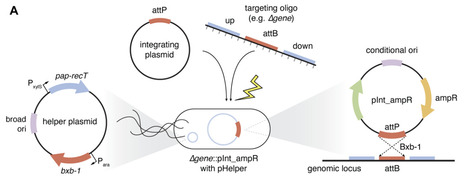
The continual advancement of genetic tools has been critical to our modern understanding of bacteria, with transposons, plasmids, and homologous recombination becoming workhorses of molecular microbiology. However, precisely specified reverse genetic approaches remain painstakingly slow and inaccessible, particularly in non-model strains. This reality is exemplified by the opportunistic pathogen, Pseudomonas aeruginosa (Pa), where conventional allelic exchange remains the dominant reverse genetic method. Here, we adapt a rapid genetic toolkit for use in Pa, relying directly on commercially available oligonucleotides (120 bases) to create precise genomic mutations through homologous recombination (i.e. oligo recombineering). Oligo Recombineering followed by Bxb-1 Integrase Targeting (ORBIT) uses a short attachment site for an integrating plasmid, which provides traditional antibiotic selection and can also carry flexible cargo. We establish Pa ORBIT works effectively for gene deletion without off target mutations, optimize protocol parameters (e.g. oligo length, electroporation), and demonstrate markerless and clean deletions. Importantly, our toolkit works well in clinical Pa strains as demonstrated by constructing efflux pump deletions in three different isolates. To test the high throughput capabilities of Pa ORBIT, we created over 160 degron-based hypomorphs (i.e. knockdowns) across 43 essential proteins in a pooled mutant library. Upon screening this library with and without antibiotics, we identify highly vulnerable essential proteins and hypomorphs that display synergy with clinical drugs. Therefore, ORBIT can be used for cutting edge low and high throughput investigations in this priority pathogen, setting the stage for answering critical basic and clinical science questions.
|
|
Scooped by
mhryu@live.com
Today, 12:26 PM
|
Stability is a desirable property for agricultural microbiomes, but there is a poor understanding of the mechanisms that mediate microbial community stability. A representative bacterial synthetic community from maize roots has been proposed by Niu et al. (2017, PNAS, 114:E2450) as a model system to study microbiome stability. This SynCom assembles stably when all seven members are present, but community diversity collapses without the keystone E. ludwigii strain. In this study, we used complementary in vitro experiments and in silico metabolic modelling to assess the role of metabolites for the stability of this SynCom, by defining the metabolic niches occupied by each strain, as well as their cross-feeding phenotypes and B-vitamin dependencies. We show that the individual member strains occupy complementary metabolic niches, measured by the depletion of distinct metabolites in exometabolomic experiments, as well as contrasting growth phenotypes on diverse carbon substrates, patterns which are largely recapitulated by computational simulations. Minimal medium experiments show that the established seven-member community comprises a mixture of prototrophic and auxotrophic strains. Correspondingly, experimental and in silico cross-feeding phenotypes showed that spent media harvested from the prototrophic strains can sustain growth of two auxotrophs and let to the identification of B-vitamin dependencies. Altogether, this study highlights the complementary power of in vitro and in silico approaches and suggests that the metabolic mechanisms of this SynCom can serve as design principles to inform the rational assembly of stable plant-associated microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:16 PM
|
Extracellular pH is a key microenvironmental factor shaping cell physiology and disease, creating a need for quantitative biosensors that can capture dynamic changes in pHe at the surface of individual living cells. Here, we develop a genetically encoded, ratiometric extracellular pH biosensor through systematic screening of a modular library of membrane-display designs that combine SEpHluorin with a pH-stable reference fluorophore. Screening identified a cell-surface-localised mKate2-SEpHluorin construct, named SurpHer, that exhibits dynamic ratiometric responses across the pHe range of 6 - 7.8. SurpHer shows robust membrane localization and extracellular pH responsiveness across diverse human cell types including HEK293T, PANC-1 and MDA-MB231 cells. Following stable integration in MDA-MB-231 cells, SurpHer enabled time-course imaging of pHe gradients in a microfluidic platform for modelling tumor microenvironments. SurpHer enables real-time interrogation of the pericellular pH environment of tumor cells and, more broadly, provides a strategy to probe microenvironmental pH dynamics across diverse biological contexts.
|
|
Scooped by
mhryu@live.com
Today, 12:04 PM
|
Accurate monitoring of canopy nitrogen content is essential for sustainable nitrogen management, yield improvement, and environmental protection in industrial maize production. However, the high dimensionality of hyperspectral data and the limited accuracy and interpretability of existing models hinder practical applications. This study was conducted in Heilongjiang Province, China, using the maize cultivar Jinboshi. Genetic Algorithm (GA), Successive Projections Algorithm (SPA), and their hybrid strategy were compared for spectral band optimization. Sensitive vegetation indices were selected using multiple evaluation criteria, and a 0–2 order fractional-order derivative (FOD) method was applied to construct optimal two-dimensional (2D) and three-dimensional (3D) spectral indices. A stacked ensemble learning model was developed using XGBoost, GBDT, and Ridge as base learners and Bayesian Ridge as the meta-learner. Interpretability techniques were applied to analyze feature contributions. The GA–SPA hybrid strategy effectively improved key spectral band selection. The 3D spectral index based on FOD achieved superior performance compared to vegetation indices and 2D indices (R2p = 0.801, RMSEP = 0.481). The optimized multi-source feature set combined with the stacked ensemble model yielded the best performance (R2p = 0.826, RMSEP = 0.450). Features from the red-edge and near-infrared regions, along with the 3D index, were the primary contributors to model predictions, consistent with plant nitrogen physiology. The proposed framework, integrating feature optimization, advanced modeling, and interpretability analysis, provides an effective tool for precise nitrogen management in industrial maize and supports improved production efficiency with reduced environmental impact.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Language models trained on biological sequences are advancing inference tasks from the scale of single proteins to that of genomic neighborhoods. Here, we introduce ProteomeLM, a transformer-based language model that uniquely operates on entire proteomes from species spanning the tree of life. ProteomeLM is trained to reconstruct masked protein embeddings using the whole proteomic context, yielding contextualized protein representations that reflect proteome-scale functional constraints. Notably, ProteomeLM’s attention coefficients encode protein–protein interactions (PPI), despite being trained without interaction labels. Furthermore, it enables interactome-wide PPI screening that is substantially more accurate, and orders of magnitude faster, than amino acid coevolution-based methods. We further develop ProteomeLM-PPI, a supervised model that combines ProteomeLM embeddings and attention coefficients to achieve state-of-the-art PPI prediction across benchmarks and species. Finally, we introduce ProteomeLM-Ess, a supervised gene essentiality predictor that generalizes across diverse taxa. Our results demonstrate the potential of proteome-scale language models for addressing function and interactions at the organism level.
|
|
Scooped by
mhryu@live.com
Today, 12:08 AM
|
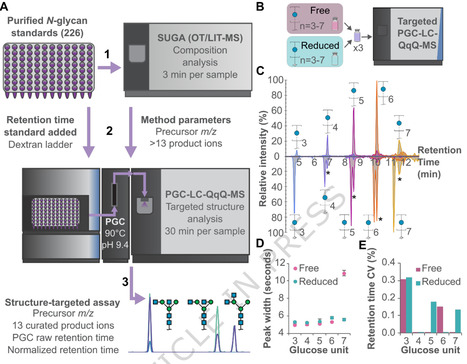
Measurements of glycans modifying glycoproteins are hampered by the lack of standards that reflect the wide diversity in structure typically observed. To this end we exploit a large library of N-glycan standards comprised of a unique collection of 226 N-glycans including oligomannose, hybrid, and complex-type and apply a method employing porous graphitised carbon (PGC) and liquid chromatography mass spectrometry (PGC-LC-MS) to provide a high-resolution separation and characterization of underivatized N-glycan structures. Chromatogram libraries arising from this study include retention time data, diagnostic fragments, and validated structural assignments, providing a robust platform for both targeted and discovery-based glycomics. Here we establish this generated data as an N-glycopedia, the resource in which researchers can compare this collective data to N-glycans under study and overcome the limitations of only having compositional data and predicted structures. The technology is easily expandable to include additional N-glycans as new standards become available. Researchers created N-glycopedia, a reference library of 226 sugar molecules found on proteins, enabling a native glycomics method to precisely identify and measure these sugars, which influence immunity and disease, without chemical pre-treatment.
|
|
Scooped by
mhryu@live.com
May 20, 11:50 PM
|
Plastic waste such as polyethylene terephthalate (PET) is a major environmental burden, and enzymes capable of degrading PET are emerging as biocatalytic tools for sustainable recycling. Progress in improving PET hydrolases (or PETases) has been constrained by the lack of simple and reliable screening systems. Here, we report a functional screen for PETase activity in E. coli based on a zone-clearing assay, where the YebF secretory pathway is used to secrete YebF-PETase onto agar plates supplemented with bis(2‑hydroxyethyl) terephthalate (BHET). Enzyme activity is observed as zones of clearance around E. coli colonies that express active YebF-PETases, as insoluble BHET is converted to soluble products. As proof of concept, the screen was used to evaluated libraries of YebF-LCC-PETase generated by site‑saturation mutagenesis at the active site residues Y95, L102, and V212. This led to the identification of the more active LCC-PETase variants V212T and L102F‑V212T. The secretion‑based assay was then validated using turbidity assays and untagged LCC‑PETase variants, where the L102F‑V212T variant was confirmed to be more active against PET than the wild type enzyme. Docking simulations indicated that V212T improves substrate positioning in the active site while L102F modifies surface charge and hydrophobicity, potentially enhancing binding to the hydrophobic substrate. Overall, the YebF secretion‑driven functional screen serves as a straightforward platform for identifying improved PETase variants and potentially other plastic degrading enzymes.
|
|
Scooped by
mhryu@live.com
May 20, 5:10 PM
|
This investigation revealed that the application of specific microbial inoculants could facilitate the effective biodegradation of polyethylene terephthalate (PET) microplastics collected from urban garbage sites, resulting in non-toxic end products. We employed Azotobacter chroococcum (MTCC 3853), Rhizobium leguminosarum (MTCC 9766), Azospirillum brasilense (MTCC 4036), and Trichoderma viride (MTCC 9681) for PET microplastics degradation and assessed their degradation efficacy through a series of controlled in vitro batch experiments. The study encompassed quantitative analysis of PET weight loss, detailed chemical profiling of degradation intermediates and products, biofilm formation assessment, microbial growth monitoring, and measurement of plastic-degrading enzyme induction. To comprehensively evaluate environmental safety, phytotoxicity assays were performed on Vigna mungo and Vigna radiata, while zebrafish embryos and adults were subjected to acute and embryonic toxicity tests. A. chroococcum (MTCC 3853) was identified as the most efficient strain, showing the greatest reduction in PET mass, enhanced biofilm formation, sustained microbial growth, and peak enzymatic activity, with no detrimental effects on plant or aquatic models, confirming the safety of the biodegradation process. These results underscore the potential of A. chroococcum (MTCC 3853) as a powerful and environmentally friendly solution for microplastic remediation in urban environments.
|
|
Scooped by
mhryu@live.com
May 20, 5:04 PM
|
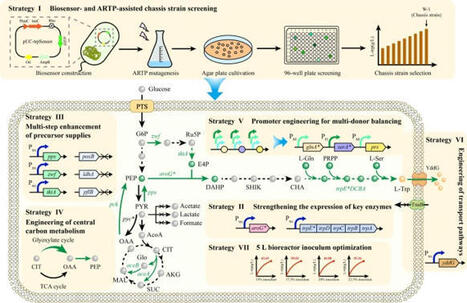
L-Tryptophan is an important aromatic amino acid with wide applications across the food, pharmaceutical, and feed industries. However, its efficient microbial production remains challenging due to complex metabolic networks and multi-level feedback regulation. In this study, we constructed a highly efficient E. coli cell factory for L-tryptophan biosynthesis by combining systematic metabolic engineering with high-throughput screening. Initially, a tnaC-based biosensor was developed and coupled with atmospheric and room temperature plasma (ARTP) mutagenesis to isolate high-performance chassis strains. Central carbon metabolism was subsequently reprogrammed to minimize carbon loss and channel metabolic fluxes toward essential precursors, phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P). To further alleviate pathway bottlenecks, promoter engineering was utilized to balance the intracellular supplies of L-glutamine, L-serine, and phosphoribosyl pyrophosphate (PRPP). This targeted intervention yielded a 21.61% increase in L-tryptophan accumulation. Product transport systems were then engineered to enhance extracellular secretion and mitigate intracellular toxicity. Following the optimization of inoculum size and feeding strategies in a 5 L bioreactor, the final engineered strain (W-24) produced 50.83 g/L of L-tryptophan within 40 h, achieving a yield of 0.185 g/g glucose. This multi-modular engineering framework establishes a robust platform for L-tryptophan biosynthesis and provides a scalable strategy for the industrial production of other valuable aromatic compounds.
|
|
Scooped by
mhryu@live.com
May 20, 4:58 PM
|
Plant pathogens possess about twice as many chemoreceptors as the bacterial average, suggesting broad chemotactic capacities. The signals recognized by most phytopathogen chemoreceptors are unknown, and the reasons for this elevated chemoreceptor number is unclear. We identified the signals recognized by three chemoreceptors, PacH, PacI and PacG, in the global phytopathogen Pectobacterium atrosepticum. The ligand-binding domains (LBDs) of these chemoreceptors share modest sequence similarity, but the signals they recognize are structurally similar, and their biosynthetic pathways are interwoven. Whereas PacH and PacI recognized benzoate derivatives, including salicylate, vanillin and p-hydroxybenzoate, PacG bound agmatine, feruloylagmatine and p-coumaroylagmatine. These compounds are known plant defense compounds, their production is induced by pathogen attack, and they typically accumulate at infection sites. All compounds, except agmatine, induced chemoattraction, which was abolished by mutations in the corresponding genes. Agmatine competed with feruloylagmatine and p-coumaroylagmatine for PacG-LBD binding in vitro and antagonized chemotaxis in vivo. A mutant in pacG, but not in other chemoreceptor genes, showed reduced virulence in planta. We report high-resolution structures of PacG-LBD that were used for ligand-docking experiments to identify its binding pocket. PacH, PacI and PacG homologs were identified in other important phytopathogens belonging to the Burkholderia, Erwinia, Ralstonia, Pectobacterium and Dickeya genera. This is the first report of chemotaxis to feruloylagmatine, p-coumaroylagmatine and p-methoxybenzoate, expanding the range of chemoeffectors. Bacteria thus exploit plant defense responses by moving to compounds that are secreted at infection sites in response to pathogen attack. Chemotaxis to plant defense compounds may be a means to access infected plants and infection sites.
|
|
Scooped by
mhryu@live.com
May 20, 4:43 PM
|
The performance of prime-editing (PE) systems has been improved by systematic engineering of their protein and small RNA components but the structured RNA motifs appended to the 3′ end of PE guide RNAs (pegRNAs)—a key determinant of pegRNA stability and editing efficiency—have not been extensively studied. We introduce PE-PRISM, a high-throughput pooled screen to identify and optimize these 3′ RNA motifs in human cells. Here, using PE-PRISM, we evaluated 2,858 RNA motifs across four iterative libraries, including natural and engineered pseudoknots, G-quadruplexes and reverse transcriptase recruitment elements. We applied structure-guided mutagenesis and combinatorial variant screening to refine hits, culminating in the engineered and evolved pseudoknot variants tevo2.0, eHAV and eSBRMV1-A. In a screen correcting 847 pathogenic ClinVar variants, the top-performing motifs improved PE efficiency over the widely used tevopreQ1 motif for >90% of edits. They also increased PE efficiencies for correcting disease-associated mutations in primary human cells and in vivo in mouse brain and liver. Prime editing is improved with engineered RNA-stabilizing motifs.
|
|
Scooped by
mhryu@live.com
May 20, 4:32 PM
|
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are natural products with diverse structures and functions. Here, we report the discovery of a family of RiPPs whose biosynthetic gene clusters are widespread in the Bacillota genomes and often co-localize with those of lasso peptides, another distinct family of RiPPs. The synthesis of both kinds of RiPPs relies on specific interactions between small adapter protein domains known as RiPP recognition elements (RREs) with their precursor peptides. As these latter share a conserved RRE-binding motif, conflicts between the two biosynthetic pathways may emerge. Through biochemical and structural studies, we reveal how the two RiPP biosynthetic systems evolved to discriminate between their cognate precursors and leader peptidases, allowing them to coexist within a single host. Thus, our study provides insights into the evolutionary diversification of RiPP families. One bacterium can produce many ribosomally synthesized and post-translationally modified peptides (RiPPs) from different families. Here, the authors show how two closely related RiPP families in Bacillota co-evolved to exclude non-cognate precursor peptides, preventing biosynthetic pathway interference.
|