 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
Protein-protein binding affinity is important for understanding protein interactions within a protein complex and for identifying strong drug-peptide binders to a target protein. Many structure-based models were built previously with reasonable performance. However, such models require protein complex structure as input, which is usually unavailable due to high cost and experimental constraints. To tackle such an issue, the sequence-based CrossAffinity model was constructed in this study, using the cross-attention module to extract contextual information of interacting protein components while separating the protein complex into two distinct parts to predict the protein-protein binding affinity. CrossAffinity managed to outperform all structure-based models and sequence-based models in an S34 test set containing newer protein complex structures and binding affinity values in a timeline while being trained on an older dataset, showing generalisability to new data points. In other test sets, namely S90, S90 subset and S79*, CrossAffinity also managed to outperform all other sequence-based models while maintaining comparable performance to many recently published structure-based models. The acceptable performance and quick inference of CrossAffinity enable it to be deployed in situations requiring the prediction of the binding affinity of many protein complexes that lack structural information.
|
|
Scooped by
mhryu@live.com
Today, 1:05 AM
|
Modifying the six-nucleotide Shine–Dalgarno (SD) core motif inside the ribosome binding site (RBS) constitutes a straightforward approach for tuning bacterial translation. However, existing methods for adjusting the effective translation rate (ETR) lack predictability. Even single-nucleotide substitutions can induce substantial alterations in translation efficiency. Moreover, this unpredictability is exacerbated by variations in the leader sequence, spacer region, or coding context. By focusing on the SD core as a key, experimentally tunable determinant of translation initiation in E. coli, we introduce a coarse-grained framework that organizes SD core variants into activity clouds with consistent expression levels. This representation converts a dense sequence-to-phenotype map into an interpretable design space for coarse-grained tuning of expression. In contrast to thermodynamic tools such as the RBS Calculator, which estimate initiation from biophysical parameters, our approach is data-driven and emphasizes (i) interpretable rules over nucleotide positions, (ii) a bidirectional workflow (core → expected ETR range; target ETR → candidate cores), and (iii) simple paths between clouds that suggest minimal sequence edits. Designed with the needs of research teams in mind, our workflow prioritizes fixing the SD core first (i.e., selecting an appropriate activity cloud) to substantially narrow the spread of observed ETRs across constructs. This dampens variability introduced by flanking DNA/RNA context (leader, spacer, local secondary structure), so that subsequent fine-tuning is simpler, cheaper, and more predictable. We validate cloud stability and predictive utility using an independent high-throughput data set. Our approach provides a solid foundation for fast, interpretable coarse control of expression, while fine-grained tuning can then be achieved through flanking-region edits that account for spacing and local structure. Finally, we provide an open web interface and repository, allowing researchers to explore the hierarchy, inspect positional influences, and export candidate cores. Together, these contributions advance the Bonde et al. data set from a static lookup into a portable, actionable map for SD core guided tuning of translation in E. coli, and outline a path to extend the idea to a fully functional framework, with possible applications also to other bacteria.
|
|
Scooped by
mhryu@live.com
Today, 12:49 AM
|
Cells rely on surface receptors to sense and initiate signalling cascades essential for numerous cellular processes, but engineering of synthetic genetic circuits to sense and rewire receptor activities for user-defined cellular functions remains a challenge. Here we report a synthetic receptor-signalling induced transcription (RESIT) circuit that enables sensing and reprogramming membrane-localized receptor activation to pre-defined transcriptional programs. The RESIT system is designed based on receptor activation mediated split protease complementation and release of membrane-tethered synthetic transcriptional modules. We show that RESIT design is generally applicable to different transcriptional factors and various split viral proteases. This system is further engineered to probe Ca2+ entry accompanied with PIEZO1 induction and T cell activation, to detect oncogenic receptor tyrosine kinase (RTK) activities and to assess Ras activation proximal to plasma membranes. The versatility of RESIT system is repurposed to actuate diverse therapeutic functions including apoptosis induction, target protein degradation and T cell activation in cells with high RTK activities. The modularity and versatility of RESIT highlight its promise for interrogating juxtamembrane biochemical signaling and rewiring receptor activation to therapeutic functions.
|
|
Scooped by
mhryu@live.com
Today, 12:47 AM
|
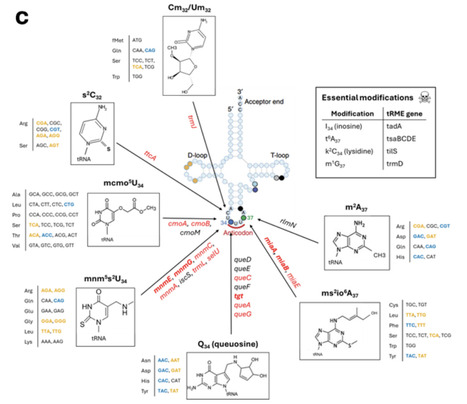
Pathogens must express virulence programs while competing for limited resources and adapting to nutrient shifts. Many virulence loci exhibit atypical codon usage, but the functional consequences of this bias remain unclear. Here, we mapped unexpected rich landscapes of diverse, functionally distinct codon usage patterns across the genomes of diverse bacterial species. In Salmonella, virulence genes formed a unique signature enriched for rare tRNAs and favoring wobble decoding. This alleviated competition with highly expressed genes, at the cost of increased dependence on specific tRNA modifications for efficient and accurate decoding. In a Salmonella systemic mouse infection model, loss of i6A37 and mnm5s2U34 modifications (miaA and mnmEG mutants) abolished virulence and selectively suppressed virulence proteins. Crucially, using recoded fluorescent proteins, we showed that virulence codon bias, by avoiding codons with high turnover, sustained increased translation rates during amino-acid starvation. Together, these findings indicate that virulence codon bias is conserved across pathogens, ensures robust expression during starvation and couples tRNA modifications to pathogenic functions, highlighting tRNA modification pathways as potential targets for broad anti-virulence strategies.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
To protect themselves against phage infection, bacteria employ diverse defense systems that are typically activated specifically upon infection. However, the mechanisms of activation and self versus non-self discrimination for most systems remain poorly understood. Here, we show that the bacterial immunity protein DARNA, once activated, cleaves a subset of host tRNAs, thereby inhibiting phage propagation. Although phages escape DARNA-mediated defense through mutations in the gene encoding single-stranded DNA-binding protein (SSB), we find that phage SSBs do not directly stimulate DARNA. Instead, DARNA is activated by single-stranded DNA presented by phage SSB, but not by the host SSB. The recognition of an endogenous nucleic acid signal promoted by a viral protein ensures that DARNA can detect and respond to a broad range of viruses while avoiding auto-immunity.
|
|
Scooped by
mhryu@live.com
Today, 12:24 AM
|
Baculovirus, an insect virus commonly used for recombinant protein expression in insect cells and gene delivery in mammalian systems, is often generated through bacmid-based engineering. To enable flexible and programmable bacmid engineering, we developed SHOT 2.0, an optimized CRISPR-associated transposon platform that mediates RNA-guided and customized bacmid editing in E. coli. The edited bacmid can be transfected into insect cells to produce recombinant baculoviruses. SHOT 2.0 supported site-specific integration of large DNA cargos (at least 14 kb) into defined loci such as v-cath and ODVe56, with integration at ODVe56 markedly improving transgene stability during serial virus passaging. The system is fully compatible with the Bac-to-Bac® workflow, enabling dual-gene insertion into the bacmid and derived baculovirus. Leveraging this platform, we constructed an all-in-one baculovirus encoding the PE5max prime editor. This vector-mediated prime editing achieves efficiencies up to 85.6% in HEK293T cells and achieves robust prime editing in hard-to-transfect cell types, including iPSCs and liver cancer cells, with efficiencies up to 37.1%. These results demonstrate that SHOT 2.0 substantially expands the baculovirus engineering toolbox, providing a flexible platform for genome editing and future gene delivery.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
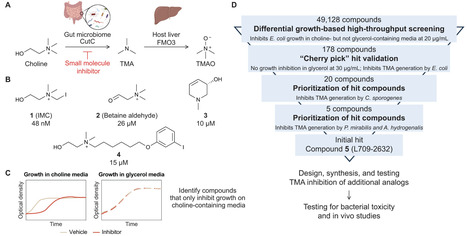
Anaerobic metabolism of dietary choline to trimethylamine (TMA) by the human gut microbiome is a disease-associated pathway. The host’s impaired ability to oxidize TMA to trimethylamine-N-oxide (TMAO) results in trimethylaminuria (TMAU), while elevated serum TMAO levels have been positively correlated with cardiometabolic disease. Small molecule inhibition of gut bacterial choline metabolism attenuates the development of disease in mice, highlighting the therapeutic potential of modulating this metabolism. Inhibitors previously developed to target this pathway are often designed to mimic choline, the substrate of the key TMA-generating enzyme choline trimethylamine-lyase (CutC). Here, we use a growth-based phenotypic high-throughput screen and medicinal chemistry to identify distinct chemical scaffolds that can modulate anaerobic microbial choline metabolism and lower TMAO levels in vivo. These results illustrate the potential of using phenotypic screening to rapidly discover new inhibitors of gut microbial metabolic activities.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
The enteric microbiome and nutrient sensing within the small intestine play critical roles in maintaining host metabolic homeostasis. Although various bacteria and some fungi have established functions in nutrient metabolism, the role of the enteric virome remains poorly understood. Here, we demonstrate that the enteric virome significantly influences carbohydrate digestion and absorption independently of the bacteriome. Furthermore, the virome elicits distinct responses across different intestinal cell types. Specifically, it activates programs for carbohydrate digestion and absorption in intestinal epithelial cells while simultaneously stimulating antigen-presenting cells―Th17 cells―to produce interleukin-22, a cytokine that curbs excessive carbohydrate uptake. The virome’s effect on carbohydrate digestion and absorption—whether suppressive or stimulatory—depends on the presence or absence of immune surveillance. This intricate interplay between metabolic and immune pathways establishes the enteric virome as a pivotal regulator of metabolism and reveals the virome’s intrinsic capacity to autonomously modulate vertebrate intestinal physiology.
|
|
Scooped by
mhryu@live.com
February 23, 11:18 PM
|
Trends in clean label food and the growing popularity of alcohol-free beers have led to a demand for natural compounds to prevent microbial spoilage. Oxidative coupling products of hydroxycinnamic acids and their amides, i.e. phenolamides, obtained via chemo-enzymatic synthesis and from barley rootlets, a beer brewing byproduct, were tested for their inhibitory activity against the beer spoilage yeast Saccharomyces cerevisiae subsp. diastaticus. Hydroxycinnamic acids, their dimers, and hydroxycinnamoylagmatines (HCAgms) did not inhibit the yeast. However, inhibitory activity was observed with HCAgm dimers. In particular, CouAgm-4-O-7′/3-8′-DCouAgm and FerAgm-4-O-7′/3-8′-DFerAgm were active at 46–255 μg/mL in beer. Notably, the newly synthesized agmatine amide of poacic acid, a stilbenoid dimer of ferulic acid, showed increased antiyeast activity compared to its monomeric precursors. These results demonstrate that amidation and oxidative coupling can increase the antiyeast activity of hydroxycinnamic acid derivatives, and that hydroxycinnamoylagmatine dimers possess potential as preservatives against wild yeast spoilage in beer.
|
|
Scooped by
mhryu@live.com
February 23, 11:00 PM
|
Keratinous waste, a major by-product of agriculture and animal husbandry, is produced in massive quantities and is notoriously recalcitrant to degradation. With the expansion of the poultry and livestock industries, keratinous waste accumulation (e.g., feathers, hooves, and horns) has become a pressing environmental concern. Keratin’s highly cross-linked disulfide bond structure is resistant to breakdown by common proteases. Keratinase, a specialized protease capable of specifically degrading keratin, has emerges as a pivotal tool for the valorization of keratinous waste, demonstrating significant potential in waste management and resource recovery. This review systematically summarizes the enzymatic properties, mechanisms of action, and microbial sources of keratinases. It elaborates on innovative keratinase applications in waste valorization (including biogas production, the generation of bioactive peptides and amino acid feedstocks, and bioplastic manufacturing) and green industries (including leather and textile processing), as well as in the pharmaceutical, cosmetic, and detergent sectors. This review provides an in-depth discussion of the major challenges hindering industrial-scale keratinase application, including low heterologous expression efficiency and insufficient stability under industrial conditions. Finally, it outlines future research directions, encompassing protein engineering, artificial intelligence (AI)-assisted design, and multi-enzyme synergistic catalysis systems, aiming to offer forward-looking theoretical insights for advanced keratinase development and industrial application.
|
|
Scooped by
mhryu@live.com
February 23, 10:52 PM
|
Pesticides are synthetic agrochemicals widely used to protect crops from pests and diseases; however, their limited biodegradability and indiscriminate application pose serious risks to non-target organisms, soil fertility, human health, and overall environmental sustainability. Conventional physical and chemical remediation strategies often fall short in restoring contaminated ecosystems, highlighting the urgent need for effective and sustainable pesticide mitigation approaches. In recent years, in situ bioremediation has emerged as a promising, eco-friendly, and cost-effective strategy for pesticide degradation in agricultural soils. Under favorable conditions, microorganisms utilize pesticides as sources of carbon, sulphur, and electrons, facilitating their breakdown through diverse metabolic pathways, with enzymatic degradation playing a central role in chemical transformation. Microbial consortia exhibit enhanced degradation efficiency by leveraging functional diversity and synergistic interactions among their microbial members. For instance, a consortium comprising Azospirillum, Cloacibacterium, and Ochrobacterium achieved 100% degradation of 50 mg L−1 glyphosate within 36 h. Advances in microbiome engineering have further expanded the scope of bioremediation by enabling the targeted manipulation of microbial communities to improve degradation specificity and performance. Notably, the recombined genomes of Psathyrella candolleana and Pseudomonas putida, generated through protoplast fusion, degraded 78.98% of pentachlorophenol in contaminated water. Additionally, engineering the rhizosphere with plant growth–promoting microorganisms, combined with microbial genetic modification, has demonstrated significant potential in enhancing pesticide degradation while simultaneously improving crop growth and productivity. Such integrative approaches represent a sustainable pathway towards resilient agroecosystems. This review synthesises current knowledge on the impacts of pesticides on crop physiology and metabolism, explores conventional and advanced microbe-mediated degradation strategies, and highlights the role of microbial engineering and consortia-based systems. Furthermore, it discusses emerging technologies, environmental and economic benefits, and recent patentable innovations, underscoring their relevance for sustainable agriculture and ecological restoration.
|
|
Scooped by
mhryu@live.com
February 23, 10:14 PM
|
Transposable elements (TEs), such as retrotransposons and endogenous retroviruses, are increasingly recognized for their important roles in genome function and impact on disease development. Residing mostly in the ‘dark genome’ — the part of the genome that does not encode proteins — TEs have long been difficult to study because of their repetitive nature. Recent technological advances are helping researchers overcome this issue, allowing them to identify how TEs contribute to disease development and discover novel targets for therapeutics. Despite this progress, the field still faces challenges. How can we translate this growing understanding of the dark genome into therapeutics? We asked experts in the field for their thoughts.
|
|
Scooped by
mhryu@live.com
February 23, 9:35 PM
|
Sedimentary nitrogen loss is a major sink for nitrogen in marine systems mitigating excess nitrogen inputs, with anammox representing a critical pathway. Conventional understanding holds that anammox bacteria exclusively mediate ammonium oxidation, while their capacity for anaerobic urea oxidation (AUO) has remained largely overlooked. Here we present the direct measurements of AUO rates and contributions across China’s marginal sea sediments, using a revised 15N isotope pairing technique. AUO exhibited a lower rate, accounting for 15 ± 10% of anammox. Moreover, porewater ammonium concentration emerged as a key regulator of AUO contribution due to substrate competition. Extrapolation to a global scale suggests that AUO accounts for ~7% of total sedimentary nitrogen loss, with a disproportionately high contribution in deep-sea sediments. These findings identify AUO as a previously overlooked but globally relevant nitrogen loss pathway, underscoring the need to incorporate AUO and other anaerobic dissolved organic nitrogen transformations into marine nitrogen budgets. Globally, anaerobic urea oxidation accounted for around 7% of total sedimentary nitrogen loss, with a disproportionately high contribution in deep-sea sediment, according to direct isotopic measurements and contributions of anaerobic urea oxidation rates across China’s marginal sea sediments.
|
|
|
Scooped by
mhryu@live.com
Today, 1:08 AM
|
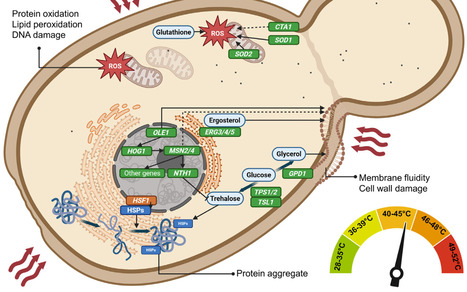
Most known yeasts are mesophilic organisms, with optimal growth at 28–30°C. However, a few species, notably Ogataea polymorpha and Kluyveromyces marxianus, can grow at temperatures around 50°C. Their thermotolerance is of both fundamental and applied interest, offering advantages for high-temperature bioprocesses. Notably, cultivation at elevated temperatures facilitates simultaneous saccharification and fermentation (SSF) of starchy and lignocellulosic substrates, reduces sterility requirements, lowers distillation costs, and enhances the rates of biochemical reactions. These features make thermotolerant yeasts attractive platforms for producing biofuels, proteins, and other valuable compounds. Despite their potential, the underlying mechanisms of thermotolerance in these species remain underexplored. Herein, we systematically review the molecular, cellular, and metabolic mechanisms underlying yeast thermotolerance and discuss their implications for high-temperature industrial biotechnology.
|
|
Scooped by
mhryu@live.com
Today, 12:57 AM
|
Chlamydomonas reinhardtii is a model green microalga that has great industrial potential as a sustainable bio-factory for recombinant protein and high-value chemical production. Efficient genome editing tools are required to redesign this organism for synthetic biology applications. CRISPR-Cas editing technologies have already been adapted for gene knockout, transgene knock-in, and precise gene editing in C. reinhardtii. However, the efficacy of CRISPR/Cas-mediated gene knockout (KO) is low, which hampers pathway engineering and functional genomic studies. Here we report that co-delivery of CRISPR-Cas gene editing reagents with short double-stranded non-homologous oligodeoxynucleotides (dsNHO) increases gene knockout efficacy up to 100-fold in C. reinhardtii. This phenomenon, referred to as non-homologous oligonucleotide enhancement (NOE), is heavily affected by the length, structure, and chemical modifications of dsNHO, and is largely mediated by the DNA double-stranded break sensor KU70/80 (KU) heterodimer in a Cas nuclease-, locus-, and strain-independent manner. Our data suggest that dsNHOs disrupt the cell's double-stranded break (DSB) sensing pathways, consequently shifting the balance of DNA repair from canonical non-homologous end joining (c-NHEJ) towards the more error-prone, microhomology-mediated end joining (MMEJ), which could be harnessed as a strategy for improving gene KO efficiency in Chlamydomonas and beyond.
|
|
Scooped by
mhryu@live.com
Today, 12:48 AM
|
Anti-CRISPRs (Acrs) are diverse proteins or RNAs that protect invading phages and plasmids from host CRISPR-Cas immunity. Most Acrs neutralize their cognate Cas proteins via direct physical interaction. Here we describe CasPRs, a particularly widespread family of DNA-binding Acrs that recognize specific sequence motifs within cas gene coding regions, thereby blocking RNA polymerase and silencing transcription. We demonstrate that eight diverse CasPRs bind to the cas8b gene to repress the type I-B CRISPR-Cas system in its native host, Listeria seeligeri. Meanwhile, a CasPR from Streptococcus dysgalactiae silences type II-A CRISPR-Cas immunity by binding to the cas9 coding sequence. We found that one CasPR is required to inhibit CRISPR immunity during lysogeny by its host prophage. Taken together, our results indicate that members of the CasPR family have diverged to silence completely unrelated CRISPR types, and suggest transcriptional repression is a common mode of phage-mediated immune antagonism.
|
|
Scooped by
mhryu@live.com
Today, 12:42 AM
|
The global rise of antimicrobial resistance has intensified the search for new microbial metabolites from underexplored environments and taxonomic groups. Extreme and geographically isolated habitats, such as Antarctic terrestrial ecosystems, represent promising reservoirs of novel biosynthetic diversity, particularly among rare and difficult-to-cultivate actinomycetes, where chemical mediators are thought to play key roles in microbial persistence and interaction under resource-limited conditions. Here, we report the characterization of kineochelins, a previously undescribed group of siderophores produced by an Antarctic isolate, Actinokineospora sp. UV203 representing difficult to cultivate actinomycetes. Structural elucidation revealed a set of closely related congener molecules with a mixed-ligand architecture consistent with metal-chelating activity. Genome mining combined with transcriptomic analysis identified the involvement of a dedicated nonribosomal peptide synthetase-encoding biosynthetic gene cluster responsible for kineochelin production. Comparative genomic analyses showed that, although kineochelin biosynthetic genes share limited homology with those of known mixed-ligand siderophores, their biosynthetic pathways differ substantially in gene content and organization, indicating a distinct evolutionary lineage. Functional characterization of kineochelins demonstrated strong and selective iron chelation, with pronounced affinity for ferric and ferrous iron. Crude culture extracts inhibited the growth of bacterial strains isolated from the same Antarctic environment, suggesting that kineochelin-associated chemistry contributes to iron-mediated competitive interactions within native microbial communities. In addition, kineochelin-enriched fractions exhibited selective inhibitory activity against the opportunistic yeast pathogen Nakaseomyces glabratus and a clinical isolate of Saccharomyces cerevisiae associated with invasive infection. Together, these findings expand the known chemical and biosynthetic diversity of the genus Actinokineospora and demonstrate that Antarctic rare actinomycetes are a valuable source of novel natural products with potential relevance for microbial ecology and biotechnology. The ecological activities of kineochelins highlight the role of iron acquisition in shaping microbial interactions in extreme environments and underscore the biotechnological potential of metabolites derived from underexplored polar microorganisms.
|
|
Scooped by
mhryu@live.com
Today, 12:26 AM
|
Efficient and scalable isolation of specific cell populations remains a central bottleneck for genome engineering, pooled screening, and cell therapy manufacturing. Here, we present DASIT (Destabilized-nanobody Antigen Selection and Identification Tool), a protein-based circuit for antigen-specific cell selection. DASIT uses a destabilized nanobody fused to an antibiotic resistance protein. In cells expressing the target antigen, binding of the nanobody fusion to the cognate antigen stabilizes DASIT, thereby coupling the presence of an antigen to a selectable signal. We developed DASIT circuits that enable robust selection of antigen-expressing cells and show that they can be designed to target distinct antigen classes and perform across cell types. Because DASIT operates at the protein level, it supports both stable integration and transient delivery, enabling recyclable selection without permanent genomic integration of resistance markers. We demonstrate scalable, FACS-free enrichment in three challenging applications: multiplexed, logic-gated integration of landing pads in human iPSCs, high-throughput CRISPR screening, and phenotypic selection of in vitro-derived neurons at transplantation scale. By decoupling selection from vector integration, DASIT establishes an automation-compatible architecture for multistep genome engineering, high-throughput library screening and large-scale cell manufacturing.
|
|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
Intestinal pH influences microbiota composition and activity, yet its impact on microbial metabolite production remains elusive. Gut bacterial tryptophan catabolism yields metabolites with opposing health effects. Indole, a precursor of indoxyl sulfate (IS), is linked to chronic kidney disease (CKD), while indolelactic acid (ILA) and indolepropionic acid (IPA) have positive health effects. Analysis of fecal pH and tryptophan metabolites in two human cohorts revealed positive correlations between fecal pH, indole, and urinary IS, and negative correlations with ILA and IPA. Fecal indole and pH showed no correlation with fecal tryptophanase (tnaA) gene abundance. In vitro fermentations showed that low pH (5.5) inhibited indole production by E. coli, enhancing tryptophan availability for C. sporogenes to produce beneficial metabolites. Human fecal cultures confirmed pH-dependent tnaA gene repression and indole suppression. These findings highlight the role of pH as a key regulator of gut bacterial tryptophan metabolism with therapeutic relevance for CKD.
|
|
Scooped by
mhryu@live.com
Today, 12:06 AM
|
Directed evolution enables the rapid creation of biomolecules with new functions, yet linking genotype and phenotype often requires laborious cell-based workflows. Here we integrate a boosted in vitro transcription-translation system capable of protein expression from single-copy DNA with ultrahigh-throughput fluorescence-activated droplet sorting, developing a simple one-day workflow for efficient protein evolution. Using this system, we evolved SP6 RNA polymerase into a highly robust variant that functions in multiple environments, including cell-free reactions and mammalian cells. We further engineered the evolved enzyme into a proximity-dependent split RNA polymerase that converts molecular interactions into transcriptional activity with minimal background. The resulting biosensors detect diverse molecular targets in vitro, including proteins, peptides, RNAs, and molecular glues. This cell-free platform provides versatile routes for evolving diverse functional biomolecules, accelerating the development of advanced biotechnologies such as diagnostics and therapeutics.
|
|
Scooped by
mhryu@live.com
February 23, 11:52 PM
|
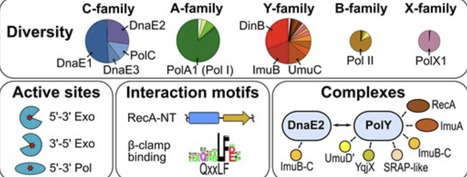
DNA polymerases are key players in DNA replication, repair, and maintenance. However, the overall abundance, diversity, and distribution of bacterial DNA polymerases have not been systematically explored. To close this knowledge gap, we computationally identified and characterized DNA polymerases and their homologs from A, B, C, X, and Y families in over 3000 representative bacterial species with complete genomes. We found that Y-family is the most abundant, followed by C and A families, whereas B and X families are rare. All species have replicative C-family polymerases, 96% have A-family polymerases, and 88% have Y-family members. In each family, we identified and annotated distinct groups, proofreading nucleases, and interaction motifs. Based on conserved associations for DnaE2 and Y-family groups, we identified 11 types of putative multimeric error-prone DNA polymerases supported by AlphaFold modeling. Approximately 90% of the complexes belong to four major types, exemplified by Meiothermus silvanus PolY–RecA complex, Mycobacterium tuberculosis ImuA–ImuB–DnaE2, E. coli Pol V (UmuC–UmuD′2–RecA), and Bacillus subtilis YqjW–YqjX–RecA. We found that distribution patterns of distinct polymerase groups and multimeric complexes are shaped by bacterial lineages, replication-system types, and environmental factors. Our results thus provide a comprehensive picture of DNA polymerase diversity and distribution across the bacterial domain.
|
|
Scooped by
mhryu@live.com
February 23, 11:15 PM
|
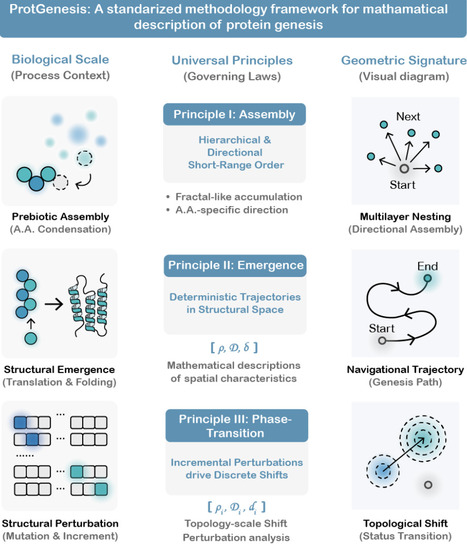
The origin of functional proteins remains a fundamental biological enigma. Although Anfinsen's dogma established sequence as the determinant of structure, and deep learning models can predict structures with high fidelity, the physical principles governing protein genesis itself, from prebiotic condensation to functional protein emergence, remain unresolved. This gap leaves a critical disconnect between mechanistic biological insights and artificial intelligence. Herein, we introduce a unified methodological framework ProtGenesis that recasts genesis of protein as a structured, deterministic navigation within a discrete structural space. We identify three universal principles governing this hierarchical organization: the Assembly Principle directs amino acids condensation into multilayer fractal-like architectures; the Emergence Principle ensures nascent peptides' emergence follow deterministic spatial trajectories; and the Phase-Transition Principle describes wherein incremental residue accrual or mutations drives precise topological phase shifts from short-range to long-range order. By quantifying these trajectories with novel tripartite spatial metrics, we reveal that protein genesis is not an abstract continuum but a principle-governed physical process with measurable coordinates. ProtGenesis thus provides an universal interpretable mathematical foundation for decoding "black-box" of deep learning models and establishes a rigorous basis for exploring, understanding, and engineering the molecular blueprint of life.
|
|
Scooped by
mhryu@live.com
February 23, 10:55 PM
|
Multi-drug resistant bacteria (MDR) pose a major public health challenge. Their ability to exchange resistance genes through Horizontal Gene Transfer (HGT) promotes the appearance of resistant strains, limiting antibiotic treatments for infections caused by these MDR bacteria. Among alternative approaches, phage therapy stands out as a promising strategy that utilizes bacteriophages to specifically target and effectively eliminate bacteria. This narrative review provides an overview of the current knowledge on the use of whole bacteriophages as antimicrobial agents in human and veterinary medicine, as well as in the food industry whether used alone, in cocktails, or combined with antimicrobials. While whole phages offer high specificity and an efficient elimination of bacteria, their application is associated with several limitations, including their contribution to HGT, the emergence of bacterial resistance, their narrow host range, the immune recognition, and the difficulties posed by their regulation. To address these challenges, this review focuses on phage-derived enzymatically active proteins, such as endolysins and depolymerases, as alternative antimicrobial tools, used alone or in combination. These phage components, being smaller and structurally simpler than whole phages, behave more similarly to conventional antimicrobial compounds. They have so far presented a low risk of bacterial resistance appearance and less chance of immune response. In addition, their classification as antimicrobial enzymes or conventional biologics could facilitate regulatory approval by aligning with existing regulatory frameworks. A total of 40 studies were included in this narrative review, highlighting the outcomes of applications involving whole bacteriophages (n = 11) and phage-derived enzymes, including endolysins and depolymerases (n = 27).
|
|
Scooped by
mhryu@live.com
February 23, 10:25 PM
|
An artificial-intelligence coach can help peer reviewers to provide more constructive and less toxic feedback, according to a new study. Whether that improves the quality of research papers, however, remains to be seen. Scientists doing peer reviews are increasingly turning to AI for a variety of tasks, including searching for relevant literature, sharpening prose and more. James Zou, a computer scientist at Stanford University in California, and his colleagues set out to assess whether large language models (LLM) could help to address a common complaint about peer reviews: feedback often lacks thoroughness or strikes the wrong tone. At the 2023 Association for Computational Linguistics annual meeting in Toronto, Canada, for example, authors of conference papers flagged 12.9% of reviews as being poor quality. That’s mainly because the reviews were vague, says Zou, with broad, simple comments such as “not novel”. Reviews can also, rarely, be unprofessional or include personal attacks, with comments such as “these authors don’t know what they’re talking about”, says Zou. Others make factual errors, for example criticizing work for omitting an analysis when that analysis is, in fact, there.
|
|
Scooped by
mhryu@live.com
February 23, 10:09 PM
|
Reactive sulfur species (RSS) are increasingly recognized as important bioactive agents across phyla. Application of newly developed chemical tools, detection methods and multi-omics techniques has uncovered new and specific roles of endogenous RSS and revealed that a number of biological actions previously attributed to reactive oxygen species or reactive nitrogen species can also be mediated by RSS. This Review describes the versatile chemical biology of RSS with focus on persulfide formation on cysteine residues. We examine their pro-oxidant and antioxidant capacities, involvement in redox signaling and metabolic pathways, stress responses, and their role in the pathophysiology of major disease groups, including cardiovascular and neurodegenerative diseases and cancer. We also provide a critical discussion of available detection methods and potential pharmacological and genetic approaches to adjusting persulfide levels. We cover current knowledge and its limitations, along with practical recommendations for advancing persulfide-based therapeutic interventions. This Review discusses current research and future directions in protein persulfidation and polysulfidation to enable their full potential in redox-based therapeutic efforts.
|




























koonin