Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:30 AM
|
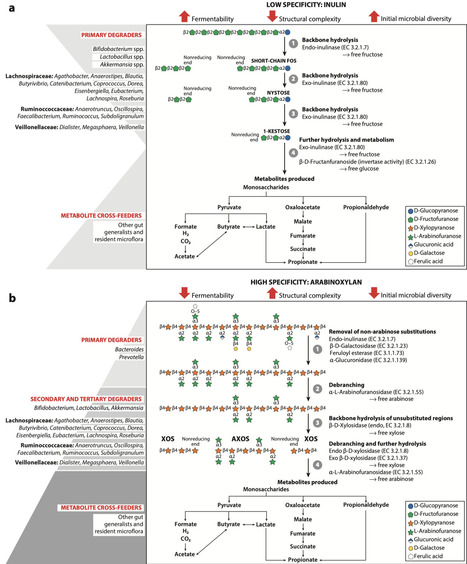
Dietary fibers are crucial in shaping gut microbial composition and functionality. Physical complexity and chemical interactions between fibers and the gut environment lead to diverse and specialized responses that involve entire food webs of gut bacteria; however, there is comparatively less emphasis on understanding ecological dynamics to predict these outcomes. These responses may potentially promote either a broader (less specific) or narrower (more specific) group of gut bacterial taxa, which may vary across individuals. This review examines fiber specificity at the organismal and community levels by exploring mechanistic interactions among dietary fibers and gut bacteria. We discuss the interplay of exogenous and endogenous factors and the structure–function relationships influencing fiber specificity. We establish a mathematical framework to describe specificity in fiber–microbiome interactions based on directionality, magnitude, and stochasticity of fiber–microbiome ecological responses. Finally, we identify research gaps to enhance fiber–microbiota predictions, with implications for strategies aimed at optimizing fiber design.
|
|
Scooped by
mhryu@live.com
Today, 1:24 AM
|
Intracellular protein patterns govern essential cellular functions by dynamically redistributing proteins between membrane-bound and cytosolic states, conserving their total numbers. This review presents a theoretical framework for understanding such patterns based on mass-conserving reaction–diffusion systems. The emergence, selection, and evolution of patterns are analyzed in terms of mass redistribution and interface motion, resulting in mesoscale laws of coarsening and wavelength selection. A geometric phase-space perspective provides a conceptual tool to link local reactive equilibria with global pattern dynamics through conserved mass fluxes. The Min protein system of E. coli provides a paradigmatic example, enabling direct comparison between theory and experiment. Successive model refinements capture both the robustness of pattern formation and the diversity of dynamic regimes observed in vivo and in vitro. The Min system thus illustrates how to extract predictive, multiscale theory from biochemical detail, providing a foundation for understanding pattern formation in more complex and synthetic systems.
|
|
Scooped by
mhryu@live.com
Today, 1:16 AM
|
Chromatin is essential for eukaryotic life. Tens of thousands of chromatin regulator (CR) proteins exist in eukaryotic genomes that are predicted to modulate chromatin states; however, their molecular functions remain largely untested experimentally. Here, we construct a library of over 300 full-length CRs from humans, plants, yeast, protozoa, and virus, each fused to DNA-binding domains, and test their direct effect on transcriptional repression and activation in plants and human cells. We discover CRs with cross-kingdom functionality when transferred across eukaryotes, including CRs that outperform existing tools for programmable transcriptional repression and activation in plants and human cells. Using pooled CRISPR screens, we demonstrate a suite of CRISPR repressors that titrate gene expression at intermediate levels. Finally, we identify RCOR1 and MTA2 as universal eukaryotic repressors that retain repressive activity in plants, yeast, and human cells. Our toolkit advances synthetic eukaryotic engineering and expands our understanding of CR functionality across eukaryotes.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
Halophiles enable convenient open, unsterile cultivation, but excessive salinity causes downstream burdens. This study engineered Halomonas bluephagenesis via genome editing, adaptive evolution, and metabolic rewiring to create a series of super low-salinity-adapted halophiles with robust growth in as low as 0.1% w/v NaCl salinity and alkaline pH 9 circumstances while preserving high-salinity resilience, significantly broadening osmotic tolerance for open, unsterile growth with contamination resistance. Multiomics and phenotypic analyses suggest mechanisms of the broad salinity-adaptive halophiles reflected in changing surface properties, enlarged morphology, auto-flocculation, and broader bioproduct synthesis. The optimized strain supported stable 7 l bioreactor cultivation at 5 g/l NaCl to reach 76 g/l cell dry weight containing up to 86% polyhydroxybutyrate (PHB) with a 39% glucose-to-PHB yield within 44 h of contamination-free growth under open, unsterile conditions, demonstrating the value of Halomonas for flexible industrial applications without high salinity concerns.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
Host-associated microbiomes often display host specificity and heritability, yet the evolutionary processes under which such structured communities first emerge are still unclear. In particular, the conditions by which intergenerational (i.e. vertical) transmission of microbes can evolve and generate host-specific microbiomes are still unresolved. Here, we present an eco-evolutionary theory of microbiome assembly under minimal assumptions of microbial dynamics (i.e. neutrally driven by environmental fluctuations) and host control. We consider the adaptive evolution of microbial and host traits, including microbiome size and vertical transmission. We show that environmental fluctuations can generate enough among-host microbial variation to enable host-level selection favoring beneficial microbiome configurations. Vertical transmission can then evolve and, even when weak, allow microbiome specificity to be inherited and amplified across generations despite continuous influx from the external environment. Selection is most effective at intermediate levels of environmental fluctuation and host lifespan, revealing fundamental trade-offs between stochastic assembly, inheritance, and dispersal of microbes. The resulting microbiomes are dense, host-specific, and heritable, yet retain high intraspecific variability and lack strict phylosymbiosis. Simulated patterns of microbial dominance, diversity, and host-microbiome dissimilarity closely match those observed in nature, as evidenced using marine sponge microbiomes. Our results provide a mechanistic theory for the early evolution of host-associated microbiomes, showing that beneficial and species-specific communities can arise through selection and inheritance prior to the evolution of dedicated host-control mechanisms.
|
|
Scooped by
mhryu@live.com
February 11, 11:48 PM
|
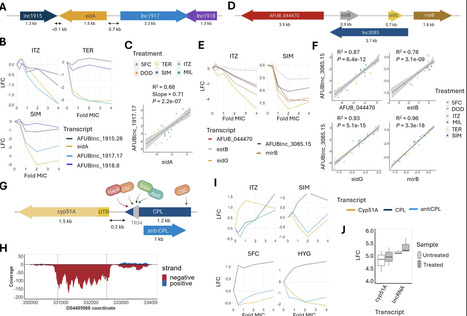
Recent data suggests one fungus, Aspergillus fumigatus, causes more deaths annually than HIV or malaria combined. Coupled with rapid emergence of antifungal drug resistance, the limited range of effective treatments, and mortality rates of >50%, aspergillosis represents a major challenge in infectious diseases. Recent studies have identified long-noncoding RNAs (lncRNAs) involved in drug resistance and virulence in pathogenic yeasts such as Candida spp. However, there is very limited knowledge of lncRNAs in human pathogenic moulds, including A. fumigatus. Here we exploit transcriptomics data of A. fumigatus exposed to different environments to annotate transcripts mapping to 2388 genomic loci. After manual curation we generate a database of over 1000 lncRNAs. We observe that the lncRNAs display orchestrated transcriptional profiles upon drug treatment and many are proximal to genes involved in azole sensitivity. We knock out a set of intergenic lncRNAs and perform a large-scale phenotypic analysis to identify 60 lncRNA mutants displaying condition-dependent fitness changes with 35 mutants exhibiting a positive growth phenotype under azole stress. Overall, this study generates and experimentally validates an important resource that will enable the wider research community to increase understanding of the functional importance of lncRNAs in A. fumigatus, including their involvement in drug sensitivity. Here, the authors present a bioinformatic pipeline to annotate lncRNA genes in Aspergillus fumigatus. The majority of these lncRNAs are only conserved at the species level, and their expression is highly responsive to antifungal exposure in Aspergillus fumigatus.
|
|
Scooped by
mhryu@live.com
February 11, 11:11 PM
|
Some legumes encode families of NCR (Nodule-Cysteine-Rich) peptides that cause their rhizobial partners to terminally differentiate during the development of a nitrogen-fixing symbiosis. Sinorhizobium meliloti, whose plant hosts Medicago truncatula and Medicago sativa express ca. 600 NCR peptides during root nodule development, possesses a symbiotically essential BacA protein that imports certain NCR peptides into the cytoplasm. This import permits proteolytic degradation of the NCR peptides, thereby protecting the endocytosed bacteria from their antimicrobial peptide-like lethality, while also allowing certain NCR peptides to undergo their symbiotically critical interactions with cytoplasmic components, for example heme-sequestration in the case of NCR247. Our study employed 54 S. meliloti bacASm missense mutants (35 to cysteine and 19 to glycine) that we tested for protein production, ability to establish a nitrogen-fixing symbiosis, and their susceptibility to killing by higher levels of the NCR247 and the Bac7(1-35) peptides. We also used the Single Cysteine Accessibility Method to make topological inferences. Our detailed genetic, biochemical, structural, and physiological analyses have revealed that BacASm and SbmA homodimers function as finely tuned transporters, whose structures can be relatively easily disrupted by single amino acid changes. Our finding that several mutations that differentially separate nitrogen-fixation, NCR247 import, and Bac7(1-35) import map to the lining of the peptide-binding cavity suggests a molecular explanation underlying the paradoxical observation that SbmA/BacAs from pathogens can fully replace BacASm, whereas BacAs from other rhizobia cannot.
|
|
Scooped by
mhryu@live.com
February 11, 4:58 PM
|
Plants and bacteria have coevolved over hundreds of millions of years, forming complex associations ranging from mutualism to pathogenicity that are essential for plant survival and ecosystem function. Bacterial adaptation to plant environments involves dynamic evolutionary mechanisms including horizontal gene transfer, gene regulation, and metabolic specialization, enabling bacteria to persist and specialize within diverse plant-associated niches. Here we review how evolutionary forces such as selection, drift, and gene flow shape bacterial genomes, regulatory networks, and ecological strategies in response to plant-imposed pressures, underpinning both beneficial and pathogenic lifestyles. Understanding these processes provides a unified evolutionary framework for bacterial adaptation to plants and highlights their implications for sustainable agriculture and microbiome-based innovations.
|
|
Scooped by
mhryu@live.com
February 11, 4:51 PM
|
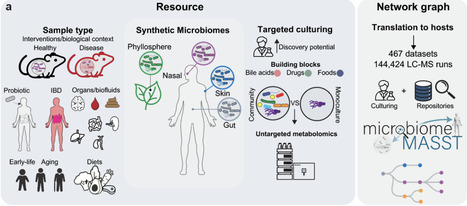
Establishing the biological context of microbial metabolites remains a major challenge. We present microbiomeMASST, a metadata-driven network graph that maps metabolites across 467 available datasets with 144,424 mass spectrometry files from humans, animals, and microbial culture systems. MicrobiomeMASST integrates monocultures, synthetic communities, and host-associated samples across multiple body sites and plants. MS/MS spectra can be queried to trace occurrence across hosts, experimental conditions, and interventions, enabling cross-study integration. We demonstrate this framework by contextualizing microbial-conjugated bile acids and interrogating microbiome-mediated drug metabolism. Screening gut bacteria revealed deprolylation of the angiotensin-converting enzyme (ACE) inhibitor prodrug enalapril. Using microbiomeMASST, we traced this metabolite across human cohorts, microbial isolates, environmental samples, and in Gorilla gorilla. Structural modeling and enzymatic assays showed that microbial deprolylation abolishes ACE inhibition, thereby inactivating its therapeutic effect. Together, microbiomeMASST links MS/MS spectra to biological context, converting isolated observations into an interpretable microbiome map for cross-study analysis.
|
|
Scooped by
mhryu@live.com
February 11, 2:38 PM
|
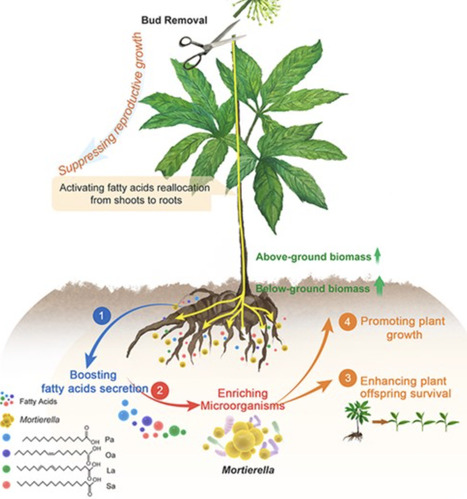
Reproductive growth suppression enhances plant productivity, yet its effects on the health and survival strategies of perennial flowering plants remain underexplored. This study investigates the trade-offs between growth and reproduction in Panax notoginseng, focusing on its relationship with rhizosphere microbiome. Through three-year, multi-site field experiments coupled with metabolomic and microbiome analyses, we demonstrate that suppressing reproductive growth significantly increases biomass, particularly in underground tissues. Mechanistically, this suppression activates fatty acid biosynthesis pathways, leading to the reallocation of fatty acid metabolites from aboveground to belowground tissues and enhancing the rhizospheric secretion of palmitic acid, oleic acid, linoleic acid, and stearic acid. These fatty acids specifically promote the growth of beneficial fungus Mortierella, which supports plant health and progeny development. Exogenous application of these fatty acids in field conditions further confirmed their role in enriching Mortierella, thereby improving plant productivity. Our findings uncover a microbial-mediated survival strategy in plants under reproductive suppression, offering mechanistic insights for sustainable rhizosphere management in perennial crops.
|
|
Scooped by
mhryu@live.com
February 11, 2:12 PM
|
Precise and tunable genetic tools are essential for high-throughput functional genomics. To address this need in the important gram-negative pathogen Pseudomonas aeruginosa, we developed and characterized a tightly regulated CRISPR-interference (CRISPRi) system that enables precise and tunable repression of essential genes. The system utilizes a rhamnose-inducible promoter to control both the Streptococcus pasteurianus-derived dCas9 and gene-specific sgRNAs, each encoded on separate plasmids for modularity and efficiency. The combination of tight regulation and high conjugation efficiency facilitated the rapid and facile construction of strains with regulated depletion of 16 essential genes spanning diverse pathways. Comparison of phenotypes across the different genetically depleted strains, including growth rate, susceptibility to antibiotics, and changes in transcriptional programs, revealed novel aspects of gene function or small-molecule mechanism of action. Finally, the rhamnose-inducible CRISPRi system supports the generation and stable maintenance of pooled mutant libraries, thereby paving the way for future genome-wide, systematic assessment of individual gene vulnerabilities, which will provide critical insights for target prioritization in antibiotic discovery efforts against this recalcitrant pathogen.
|
|
Scooped by
mhryu@live.com
February 11, 1:51 PM
|
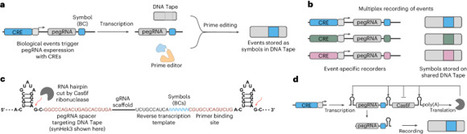
Molecular recording is an emerging paradigm for measuring biology over time. Enhancer-mediated genomic recording of activity in multiplex (ENGRAM) is a recently described synthetic biology circuit architecture that converts the transient activity of cis-regulatory elements (CREs) into stable genomic records that can be retrospectively recovered via DNA sequencing. Here we provide a step-by-step protocol for conducting ENGRAM experiments and analyzing the resulting data. We also describe key design considerations for ENGRAM recorders, summarize the strengths and limitations of ENGRAM, and highlight applications, including multiplex signal recording and high-throughput CRE screening. In contrast to other systems for DNA-based recording in mammalian systems, ENGRAM relies on prime editing-mediated insertions to record the activity of a given CRE, such that it is inherently multiplexable—for example, four-base-pair insertions can represent the activities of up to 256 distinct CREs. A further contrast lies with ENGRAM’s compatibility with DNA Typewriter, which facilitates the capture of signal order. For users with basic skills in molecular biology, mammalian cell culture and DNA sequencing analysis, ENGRAM experiments can typically be completed within 5–6 weeks. This Protocol details the use of enhancer-mediated genomic recording of activity in multiplex (ENGRAM) for molecular recording. This system converts the transient activity of cis-regulatory elements into stable genomic records that can be retrospectively recovered via DNA sequencing.
|
|
Scooped by
mhryu@live.com
February 11, 12:54 PM
|
Cell-free biosensors have the potential to become a low-cost, widely distributed technology. If deployed at scale, they can generate new large-scale data streams about human health and our environment, providing actionable information at the point of need. Here, we review the key technological advances of cell-free biosensors over the last five years and suggest a future path of technology development, including interfacing with circuits, devices, and materials that are becoming part of the next generation of biosensors. We then ask what is needed for these technologies to succeed at scale, focusing on lessons from field-deployment studies, policy, regulatory, safety, and other considerations to ensure alignment of technological developments with real-world necessities, market opportunities, and reliable use by non-experts.
|
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
Glycogen phosphorylase (GP) plays a central role in glycogen metabolism. While the structure and regulation of mammalian GPs have been extensively studied, the corresponding mechanisms in gut bacterial GPs remain poorly understood. Here, we investigate GPs from E. coli (EcGP), Segatella copri (ScGP), and Dorea longicatena (DlGP), which represent three phylogenetic clades of GPs, using enzymatic assays, cryo–electron microscopy (cryo-EM), and X-ray crystallography. We find that ScGP forms a unique pentamer that undergoes adenosine monophosphate (AMP)-dependent assembly into a dimer-of-pentamer, which inhibits activity by restricting substrate access to the catalytic site. EcGP exists in equilibrium among monomers, dimers, and tetramers, with AMP promoting tetramer dissociation and enhancing catalytic efficiency. In contrast, DlGP remains predominantly monomeric and is unresponsive to AMP. These findings uncover structural and regulatory diversity among gut bacterial GPs. Notably, the oligomeric states of GPs modulate substrate accessibility and enzyme activation, suggesting a distinct mode of allosteric regulation beyond the canonical T-to-R transition model. Because bacterial GPs contribute to the generation of glucose, their regulation may influence the composition of gut-derived metabolites that affect host glucose homeostasis and insulin sensitivity. Our study provides mechanistic insight into the structural and functional diversity of gut bacterial GPs and lays a foundation for future exploration of microbiome-mediated metabolic interactions.
|
|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
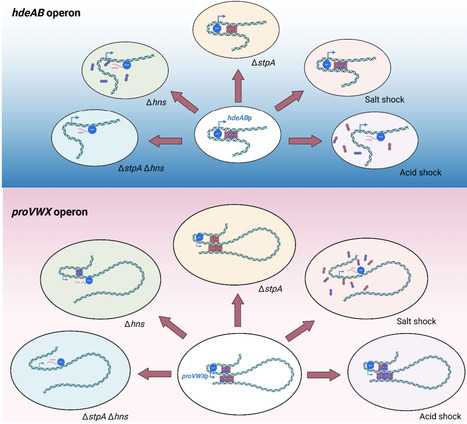
E. coli is highly sensitive to acid and osmotic stress but adapts by modulating the expression of stress responsive genes. Nucleoid-associated proteins (NAPs) play key roles in DNA organization and sensing environmental changes. The histone-like nucleoid structuring protein H-NS is an NAP acting as a global regulator of stress genes. H-NS may alter local chromatin structure to modulate the expression of such genes in response to environmental stress. The H-NS homolog StpA co-regulates several target genes, but its precise role is poorly defined. To investigate the regulatory interplay between these two proteins, we examined transcription, DNA binding and chromatin structure at two regulated operons, hdeAB and proVWX, in E. coli following exposure to acid and salt shock. Our results show that H-NS senses pH and osmotic cues to remodel chromatin and relieve repression, while StpA compensates for H-NS loss, particularly at proVWX, highlighting a coordinated regulatory network.
|
|
Scooped by
mhryu@live.com
Today, 12:40 AM
|
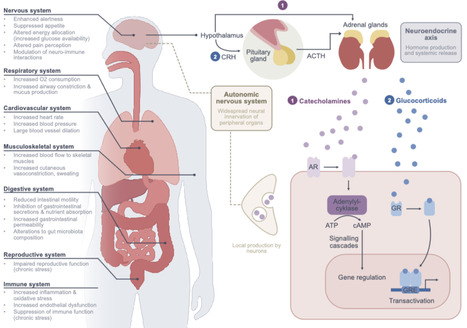
It has become increasingly appreciated that gut microbes influence host stress hormone responses through direct and indirect mechanisms. These relationships may have broad implications on hormone bioavailability, receptor signaling, and stress resilience. In this review, we summarize current evidence for microbe-stress factor interactions and their consequences for host physiology. We further examine how microbiota-stress crosstalk may contribute to inflammatory bowel disease, highlighting emerging mechanisms and potential microbiota-targeted therapies.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
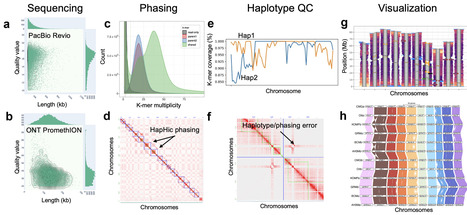
Plant genome biology is entering a new era defined by fully phased, chromosome-scale, telomere-to-telomere assemblies, enabled by the convergence of long-read sequencing technologies, improved assembly algorithms, and powerful scaffolding strategies. Gapless, haplotype-resolved genomes are now feasible even for polyploid species, shifting the bottleneck from assembly to annotation and interpretation. Genome annotation remains one of the greatest opportunities and challenges in plant biology. While ab initio methods still form the backbone of structural prediction, evidence-based frameworks that integrate RNA sequencing, chromatin accessibility, methylation, and 3D genome data are rapidly advancing the field. At the same time, artificial intelligence–driven protein-coding gene predictors are redefining ab initio gene finding, and large-scale orthology networks continue to improve functional inference. The next frontier is extending annotation beyond protein-coding genes into regulatory and structural dimensions, a goal increasingly enabled by single-cell and multi-omic technologies. Looking forward, the integration of AI, multi-omics, and large language models promises to standardize and automate workflows from DNA isolation to functional annotation. These innovations will accelerate fundamental plant biology discovery, enable next-generation biodiversity conservation, and transform strategies for crop improvement and biotechnology.
|
|
Scooped by
mhryu@live.com
Today, 12:08 AM
|
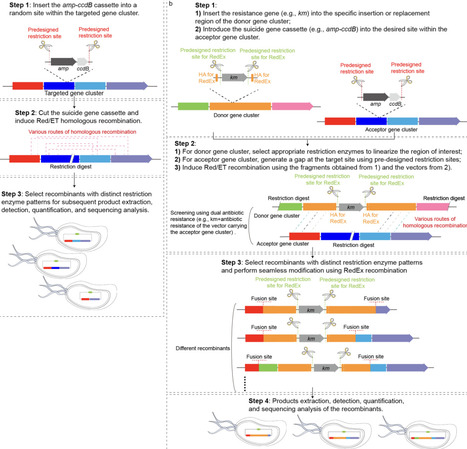
Efficient engineering of nonribosomal peptide synthetases (NRPS) is a key strategy for expanding valuable peptide natural products. Current bioinformatics-guided approaches are often constrained by a set of predefined fusion sites, remaining experimentally challenging in most NRPS systems. Here we present the Recombineering Accelerated Evolution (RACE), which harnesses Red/ET recombineering mediated partially matched homologous recombination to recapitulate recombination‑driven NRPS evolution on a highly accelerated timescale. Application of RACE to six known NRPS gene clusters generated 830 recombinants and yielded over 600 novel peptides including novel bis‑lipopeptides. These recombinants reveal 112 previously unrecognized recombination fusion sites, providing an extensive landscape for NRPS evolution and more useful resources for guiding NRPS engineering. The RACE establishes a new paradigm for programmable NRPS evolution and enables rapid discovery of bioactive peptides.
|
|
Scooped by
mhryu@live.com
February 11, 11:35 PM
|
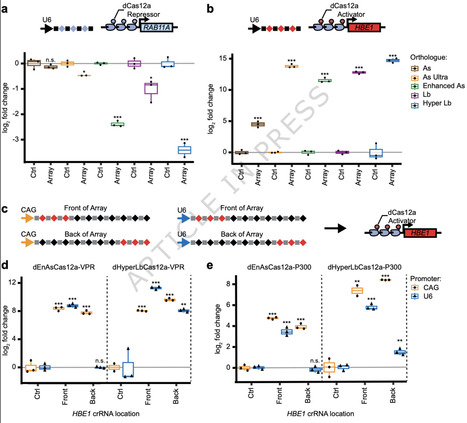
Interactions between genes or cis-regulatory elements (CREs) underlie many biological processes. High-throughput CRISPR screens have allowed researchers to assess the impact of activation or repression of gene and regulatory elements on many phenotypes. However, assessment of interactions between those genes or elements remains limited. To enable efficient highly-multiplexed control of regulatory element activity, we combine a hyper-efficient version of Lachnospiraceae bacterium dCas12a (dHyperLbCas12a) with RNA Polymerase II expression of long CRISPR RNA (crRNA) arrays. We demonstrate this system with several activation and repression domains, in cultured primary immune cells, and to differentiate induced pluripotent stem cells. We also develop approaches to use dCas12a for simultaneous activation and repression. Lastly, we demonstrate that dHyperLbCas12a effectors can be used to dissect the independent and combinatorial contributions of CREs to gene expression. These tools create possibilities for highly multiplexed control of gene expression in many biological systems. CRISPR/Cas9 screens have identified genetic contributions to many phenotypes. However, studying combinations of genes or regulatory elements remains challenging. Here, the authors use CRISPR/Cas12a to overcome those challenges and enable new approaches to study combinatorial genetic mechanisms.
|
|
Scooped by
mhryu@live.com
February 11, 5:02 PM
|
Antimicrobial resistance (AMR) is a growing global health crisis, responsible for an estimated 1.27 million deaths in 2019 alone. traditional approaches to identifying antibiotic resistance genes (ARGs) are often labour-intensive and limited in their ability to detect novel resistance mechanisms. In this study, we propose BRIDGE, a knowledge graph-based framework, to improve AMR gene prediction by integrating gene neighborhood information and protein-protein interaction networks. Focusing on Klebsiella pneumoniae and E. coli, we construct a comprehensive and biologically grounded knowledge graph using curated data from CARD, STRING, and DrugBank. We apply knowledge graph embedding models which are fed into deep neural networks to infer novel AMR links, achieving classification accuracy of up to 97%. Our results demonstrate that incorporating biologically meaningful relationships, such as gene neighbourhood information and protein interactions, enhances the predictive accuracy and interpretability of AMR link predictions. This work contributes to the development of scalable and data-integrated approaches for advancing antimicrobial resistance surveillance and drug discovery.
|
|
Scooped by
mhryu@live.com
February 11, 4:56 PM
|
Predator-prey interactions are intricately linked to ecological systems, from microorganisms to large animals. Most predator-prey studies use simplified pairwise interactions, constraining our ability to identify general principles. Here, predator prey choices are examined across scales and levels of environmental complexity. We review current knowledge and emphasize the diversity and complexity of predator-prey systems, point to challenges in integrating them, and propose a framework that could benefit predictive modelling for ecosystem functioning and resilience. To do so, we compare the tools, mechanisms and strategies deployed by micro- and macro- predators and prey defenses to show that commonalities become identifiable, and suggest structural and functional links between micro- and macro-scales. This provides arguments for both descriptive, and mathematical models. We propose that the use of microbial predators like the Bdellovibrio and like organisms (BALOs) can greatly advance the integration of experimental and mathematical modeling research, as they can provide robust empirical observations of predator-prey interactions tested under multiple conditions and levels of complexity. This facilitates model development, in turn leading to new hypotheses. We conclude by showing examples of current developments, that predator-prey interaction-based knowledge has the potential to provide novel medical tools and to improve environmental and agricultural management.
|
|
Scooped by
mhryu@live.com
February 11, 3:51 PM
|
Recent developments in DNA synthesis and sequencing allowed the construction of comprehensive gene variant libraries and their functional analysis. Achieving high-replication and thorough mutation characterization remains technically and financially challenging for long genes. Here, we developed an efficient, affordable, and scalable library construction approach that relies on low-cost DNA synthesis and standard cloning technologies, which will increase accessibility to mutational studies and help advance the field of protein science. Each degenerate codon variant is physically associated with multiple DNA barcodes during synthesis, which overcomes the need for long-read sequencing for linking variants to barcodes. We demonstrate the scalability of our approach by constructing a complete library for PDR1, a 3.2 kb multidrug resistance gene encoding a pleiotropic transcription factor in the yeast Saccharomyces cerevisiae. We demonstrate a near-perfect correspondence in the measurement of amino acid variants impact when assessed by barcode sequencing and direct sequencing of the mutated coding sequence.
|
|
Scooped by
mhryu@live.com
February 11, 2:16 PM
|
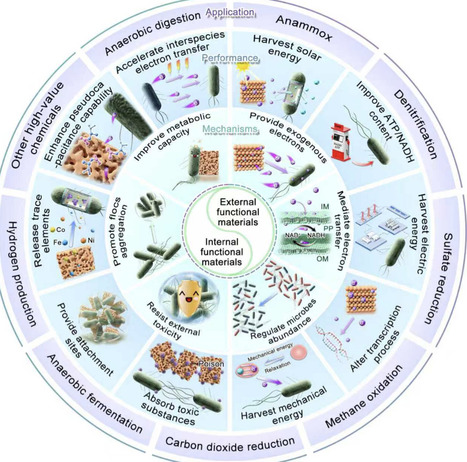
The incorporation of functional materials into anaerobic biological systems offers promising opportunities for sustainable and efficient wastewater treatment. However, there is a discrepancy between the explosive growth of emerging materials and their limited environmental applications. In this Perspective, we summarize two main functional materials that could enhance environmental biotechnology according to their energy sources. The first type, “internal functional materials”, facilitates interspecies interactions and stimulates metabolic processes. The second type, “external functional materials”, mediates the exchange with external energy. We discuss the mechanisms of enhancing interspecies and extracellular electron transfer, which largely determine the microbial metabolism of substance and energy. We reflect on the remaining challenges in bringing functional materials to practical applications and clarify the potential suitable scenarios based on the properties of these materials. In the future, designing new functional materials using artificial-intelligence-driven models should necessitate identifying specific microbial metabolic mechanisms. This Perspective highlights innovative use of internal and external functional materials to boost microbial activity and electron transfer. It also provides an intuitive interpretation for the rational design of functional materials to address the urgent need for materials in large-scale engineering applications.
|
|
Scooped by
mhryu@live.com
February 11, 2:03 PM
|
Horizontal transfer of transposable elements (TEs) is widespread in eukaryotes, driving genetic variation and often associated with bursts of TE activity. Here, we report a recent TE burst in the insect-pathogenic fungus Metarhizium anisopliae. The actively transposing TEs were likely introduced via hitchhiking on a so-called Starship, a class of large, horizontally transferable transposons. This TE burst likely triggered extensive structural reshuffling across all chromosomes, which was associated with loss of pathogenicity. Expanding our analysis to other fungi, we found that Starship-mediated horizontal transfer of TEs is a general phenomenon. Most (75%) of 522 reported Starships harbor TEs; many of which show evidence of a recent burst, in some cases likely starting from the TE copies on the Starship itself. A high fraction of TEs located on Starships also shows signatures of past horizontal transfer. Collectively, our results establish Starships as major vectors of horizontal TE transfer. Large mobile genetic elements known as Starships act as vehicles for transferring transposable elements (TEs) between fungi. Here, Griem-Krey et al. show that these ‘hitchhiking’ TEs can drive rapid evolution through genome reshuffling, which can alter fungal pathogenicity.
|
|
Scooped by
mhryu@live.com
February 11, 12:57 PM
|
The restoration of terrestrial ecosystems often requires the reestablishment of plant communities, but restorations often overlook microbial communities, which directly and indirectly structure plant diversity. Legacies of anthropogenic disturbance radically alter microbial diversity and, contrary to previous assumptions, the natural recovery of microbial communities is not guaranteed, even after decades. This necessitates established and novel forms of microbial restoration, such as the addition of key microbial taxa to boost plant diversity and ecosystem functioning. Here, we review why anthropogenically disturbed microbial communities need restoration, the positive outcomes of doing so for plant conservation, community dynamics, and ecosystem functioning, and best practices when taking these approaches. We also highlight knowledge gaps in this emerging field, such as the mechanisms underlying successful microbially based restoration of plant communities, how shifting climate conditions will impact global microbiomes, and the potential for off-target effects. Our goal is to shift the perspective of microbes as passive measures of restoration success to the restoration of microbial communities as a major objective and driver of restoration outcomes.
|