Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 6:31 PM
|
Optimization of flux distribution in central carbon metabolism is important to improve the microbial productivity. As the number of precursors required for synthesis differs for each target compound, optimal flux distribution also varies. A library of mutant strains with diverse flux distributions can aid in optimal strain screening. Therefore, in this study, we aimed to construct a library of Escherichia coli strains with stepwise changes in flux distribution by introducing mutations into the ribosome-binding sites of key enzyme genes on its chromosome. We focused on the flux ratios at the glucose-6-phosphate and acetyl-CoA branch points to enhance mevalonate production. Mutations were introduced into the RBS of pgi and gltA to vary the flux ratios of the two pathway branches. Furthermore, a combinatorial repression library comprising 16 strains was constructed by varying pgi and gltA expression at four levels, and a plasmid containing mevalonate synthesis genes was introduced into each strain. Batch cultures were performed to obtain strains with mevalonate titers and yields 2.4- and 3.4-fold higher than those of the parent strain. Overall, our combinatorial suppression library of pgi and gltA facilitated the effective identification of mutants with optimal metabolism for mevalonate production.
|
|
Scooped by
mhryu@live.com
Today, 6:05 PM
|
Pre-mRNA splicing is a core process in eukaryotic gene expression, and splicing dysregulation has been linked to various diseases. However, very few small molecules have been discovered that can modulate spliced mRNA formation or inhibit the splicing machinery itself. This study presents a novel high-throughput screening (HTS) platform for identifying compounds that modulate splicing. Our platform comprises a two-tiered screening approach: A primary screen measuring growth inhibition in sensitized Saccharomyces cerevisiae (yeast) strains and a secondary screen that relies on production of a fluorescent protein as a readout for splicing inhibition. Using this approach, we identified 4 small molecules that cause accumulation of unspliced pre-mRNA in vivo in yeast. In addition, cancer cells expressing a myelodysplastic syndrome-associated splicing factor mutation (SRSF2P95H) are more sensitive to one of these compounds than those expressing the wild-type version of the protein. Transcriptome analyses showed that this compound causes widespread changes in gene expression in sensitive SRSF2P95H-expressing cells. Our results demonstrate the utility of using a yeast-based HTS to identify compounds capable of changing pre-mRNA splicing outcomes.
|
|
Scooped by
mhryu@live.com
Today, 5:59 PM
|
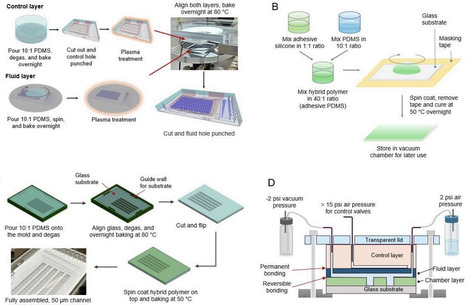
High-throughput microfluidics has transformed biomedical research by enabling precise and parallel sample handling, but most devices are single-use due to channel occlusion and contamination from experiments. Alongside low fabrication yield and reduced experimental success associated with dense microfeatures, this creates a major bottleneck for scalable high-throughput applications. We present a rapid, reusable, and modular high-throughput microfluidic platform with integrated microvalves for automation. The platform employs a multilayer architecture consisting of a custom casing, PDMS layers with dense microfeatures for fluid handling and culture, and a glass substrate. Permanent bonding is applied only between control and fluid layers, while reversible bonding is used at all other interfaces, including the substrate. Because substrate is the primary cell-contact surface and can be readily detached, the remaining layers can be disassembled, thoroughly cleaned, and reused with minimal processing on a new substrate. This approach improves repeatability and experimental success while reducing preparation time from days to ~2 hours. The disassemblable design also supports incorporation of application-specific layers between fluid layer and substrate, enhancing platform versatility for 3D culture. We validated performance through pressure/flow characterization and on-chip cell/organoid culture. Overall, our platform accelerates rapid high-throughput data generation across diverse biological applications.
|
|
Scooped by
mhryu@live.com
Today, 4:00 PM
|
Harnessing heterosis has been important to agriculture, but its full potential remains constrained by the inability to clonally propagate hybrid seeds. Synthetic apomixis, engineering asexual seed formation in sexual crops, provides a path to fix heterosis through generations. This review compiles current advances in engineering synthetic apomixis, focusing on two major components: the substitution of mitosis for meiotic divisions and maternally derived embryo development by haploid inducer or parthenogenesis. Despite rapid progress made in rice, significant challenges remain for general implementation and extension to other crops. We discuss these challenges and the possible paths forward to make synthetic apomixis feasible for widespread application in agriculture.
|
|
Scooped by
mhryu@live.com
Today, 3:47 PM
|
Understanding microbial growth and metabolism under controlled environments is critical for both fundamental research and bioprocess development. In this study, we present a cost-effective droplet-based microfluidic device enabling high-throughput screening of GFP expression in E. coli under varying glucose concentrations. Fluorinated Ethylene Propylene (FEP) tubing was selected for its low cost and compatibility with stable water-in-oil emulsification, facilitating robust droplet generation. The system achieved a 33333-fold reduction in media consumption compared to traditional Erlenmeyer flask cultures. Growth kinetics and GFP expression were assessed in both Erlenmeyer flasks and microdroplets, showing high qualitative correlation between platforms. Low glucose levels (5–10 g/L) supported rapid initial growth and early GFP production, followed by a fluorescence decline due to nutrient depletion. In contrast, higher glucose concentrations (25–50 g/L) prolonged the exponential phase and enhanced GFP production per unit biomass, though growth was slowed by overflow metabolism. In microdroplets, delayed GFP expression at 25 and 50 g/L were observed, and parallel bioreactor experiments confirmed that this delay is caused by oxygen limitation at high glucose concentrations. Importantly, the microfluidic device enables controlled variation of oxygen availability simply by adjusting droplet size or generation frequency, providing a powerful means to probe oxygen-sensitive metabolic behaviors. These results validate the microfluidic platform’s ability to mimic Erlenmeyer flask-scale dynamics, while uniquely allowing precise modulation of oxygen transfer conditions at the microscale. The system offers a reliable, miniaturized alternative for optimizing microbial bioprocesses with drastically reduced reagent use and increased experimental throughput.
|
|
Scooped by
mhryu@live.com
Today, 3:21 PM
|
Food security and environmental sustainability require innovative agricultural approaches, with plant growth-promoting microbes offering promising solutions. This study explores the growth modulation of plants by Microbacterium sp. MB15 through secreted compounds, distinguishing between the effects of total diffusible substances and volatile organic compounds (VOCs). Microbacterium sp. MB15 exhibited a dose-dependent influence on wheat and Arabidopsis germination, growth, and root architecture, shifting from stimulation at low exposure levels to inhibition at higher concentrations. Total diffusible compounds, including auxin produced by the bacterium, exert their effect on Arabidopsis through the TIR1/AFB pathway and partially require the YUCCA genes. Microbacterium sp. MB15 VOCs, comprising ethanol, acetic acid, ethyl acetate, methanethiol and dimethyldisulfide, also elicited strong modulation of seed germination and seedling growth through mechanisms only partially dependent on TIR1/AFB and YUCCA. A proteomic analysis of seedlings exposed to bacterial VOCs (ProteomeXchange # PXD069087) indicated stimulation of carbohydrate and lipid mobilization, overexpression of proteins associated with a non-canonical auxin pathway, metabolism of indolic glucosinolates, defense responses, and sulfur-redox homeostasis. Additionally, high exposure to VOCs led to repression of genes for photosynthesis and chloroplast integrity. These findings help unravel the complex molecular responses underlying plant-microbe interactions.
|
|
Scooped by
mhryu@live.com
Today, 2:26 PM
|
During the last few years, the body of data on proteins is expanding almost exponentially with the development of advanced methods for gene sequencing, protein structure determination, particularly by cryoelectron microscopy, and structure prediction using artificial intelligence-based approaches. These developments create the potential for a comprehensive exploration of the protein universe, the entirety of the proteins existing in the biosphere. Elucidation of the relationships among proteins including the most distant ones, where only the core fold is shared, is crucial for understanding protein functions, folding mechanisms, and evolution, as well as the evolution of cellular life forms and viruses. In this brief review, we discuss methods that shaped the field of protein bioinformatics, first, through comparative sequence analysis, and the recent developments in protein structure prediction that transformed the state of the art in the comparative analysis of distantly related proteins. The combination of the rapidly growing databases of genome and metagenome sequences with sensitive methods for sequence comparison and the new generation of structure analysis tools can make charting the protein universe at the structural level a realistic goal.
|
|
Scooped by
mhryu@live.com
Today, 2:09 PM
|
Pseudomonas putida is a plant-beneficial rhizobacterium that encodes multiple type-VI secretion systems (T6SS) to outcompete phytopathogens in the rhizosphere. Among its antibacterial effectors, Tke5 (a member of the BTH_I2691 protein family) is a potent pore-forming toxin that disrupts ion homeostasis without causing considerable membrane damage. Tke5 harbors an N-terminal MIX domain, which is required for T6SS-dependent secretion in other systems. Many MIX domain-containing effectors require T6SS adaptor proteins (Tap) for secretion, but their molecular mechanisms of adaptor-effector binding remain elusive. Here, we report the 2.8 Å cryo-EM structure of the Tap3-Tke5 complex of P. putida strain KT2440, providing structural and functional insights into how effector Tke5 is recruited by its cognate adaptor protein Tap3. Functional dissection shows that the α-helical region of Tke5 is sufficient to kill intoxicated bacteria, while its β-rich region likely contributes to target membrane specificity. These findings delineate a mechanism of BTH_I2691 proteins for Tap recruitment and toxin activity, contributing to our understanding of a widespread yet understudied toxin family.
|
|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
Synthetic rewriting technologies, encompassing large-scale DNA assembly, transfer, maintenance, and rearrangement, enabled de novo synthesis or large-scale modifications of genomes. While significant progress has been made in model organisms of viruses, bacteria, and unicellular eukaryotes, their development in mammalian cells faces unique challenges. This review summarizes key breakthroughs in synthetic rewriting technologies, including megabase (Mb)-scale assembly of human DNA, yeast-mediated transfer methods, bottom-up human artificial chromosomes (HACs), and genome-scale rearrangement, along with emerging applications in constructing models and decoding genomes for mammals. These tools will expand functional engineering in mammals and deepen mechanistic insights into complex biological systems. Synthetic DNA rewriting technologies enable de novo synthesis or large-scale modification of genomes. Here the authors discuss key advances in the DNA rewriting toolbox at the level of large-scale DNA assembly, transfer, maintenance, and rearrangement, whilst highlighting emerging applications.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Gene duplication has played a critical role in the evolutionary history of proteins, enabling complex multimers to emerge from simpler precursors. Yet in protein engineering, current methods for directed evolution do not exploit gene duplication, hampering access to the vast array of diverse variants that are only enriched in the presence of a wild-type copy. We establish a directed evolution strategy for multimeric proteins that harnesses gene duplication to compensate for metabolic burden and self-assembly fitness, allowing previously inaccessible variants to be enriched. Starting from a homomeric 240-mer capsid, gene duplication enables selection of both extreme homomeric variants and obligate heteromers. This strategy significantly expands engineering access to diverse high-performing variants, while also supporting a plausible model for evolutionary diversification of higher-order multimers in nature.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
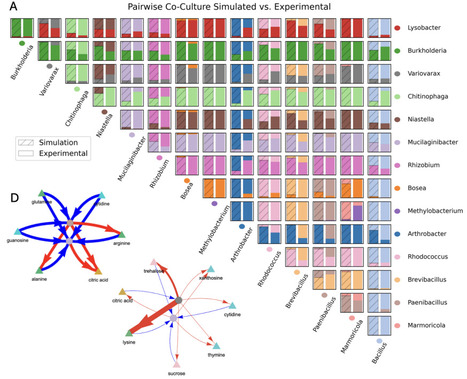
Synthetic microbial ecology aims at designing communities with desired properties based on mathematical models of individual organisms. It is unclear whether simplified models harbor enough detail to predict the composition of synthetic communities in metabolically complex environments. Here, we use longitudinal exometabolite data of monocultures for 15 rhizosphere bacteria to parametrize a consumer-resource model, which we use to predict pairwise co-cultures and higher order communities. The capacity to artificially "switch off" cross-feeding interactions in the model demonstrates their importance in ecosystem structure. Leave-one-out and leave-two-out experiments demonstrate that pairwise co-cultures do not necessarily capture inter-species interactions within larger communities and broadly highlight the nonlinearity of interactions. Finally, we demonstrate that our model can be used to identify new sub-communities of three strains with high likelihood of coexistence. Our results establish hybrid mechanistic and data-driven metabolic models as a promising and extendable framework for predicting and engineering microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:20 AM
|
Genetically encoded biosensors enable the monitoring of metabolite dynamics in living organisms. We present CoBiSe, a computational biosensor design approach using Constraint Network Analysis to identify optimal insertion sites for reporter modules in molecular recognition elements (MREs). Applied to the iron-binding protein DtxR from Corynebacterium glutamicum, CoBiSe identified a flexible connective loop (residues 138–150) for inserting the reporter module, resulting in IronSenseR, a novel ratiometric biosensor for ferrous iron (Fe2+). IronSenseR demonstrates high specificity for Fe2+ with dissociation constants of 1.78 ± 0.03 (FeSO4) and 2.90 ± 0.12 μM (FeCl2), while showing no binding to Fe3+ and other divalent cations. In vivo assessment in Escherichia coli, Pseudomonas putida, and Corynebacterium glutamicum confirmed IronSenseR’s capability to detect changes in the intracellular iron pool. The creation of IronSenseR underlines that by reducing search space and eliminating labor-intensive screening, CoBiSe streamlines biosensor development and enables precise creation of next-generation biosensors for diverse metabolites.
|
|
Scooped by
mhryu@live.com
January 12, 11:56 PM
|
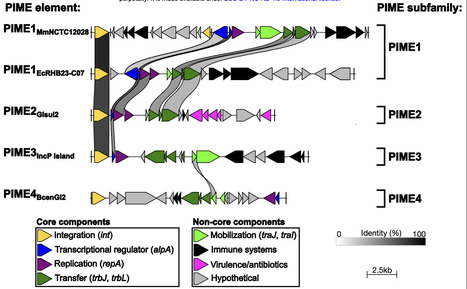
Integrative mobilizable elements (IMEs) are mobile genetic elements that reside stably integrated into chromosomes and rely on helper conjugative elements for horizontal transfer. Here, we identify and characterize a widespread family of IMEs, named Pseudomonadota Integrative Mobilizable Elements (PIMEs), which are distributed exclusively in the Pseudomonadota phylum. Genome and phylogenomic analyses reveal ∼1,000 putative PIMEs, comprising at least four distinct PIME subfamilies defined by distinctive genomic organizations and conserved hallmark features. Characterized PIMEs depend on helper conjugative plasmids of the incompatibility group P (IncP) and, upon induction, PIMEs excise, replicate and mobilize intra- and inter-species. Remarkably, the representative PIME and its helper conjugative plasmid engages in cross-complementation, revealing an unrecognized level of functional interplay between hijacker and helper element. We also demonstrate that PIMEs act as reservoirs of known and novel prokaryotic immune systems. Overall, our findings uncover an overlooked and disseminated family of IMEs, which likely plays an important role in bacterial ecology and evolution.
|
|
|
Scooped by
mhryu@live.com
Today, 6:30 PM
|
The Antarctic ecosystems are a combination of conditions, including extremely low values of temperature and humidity. Nonetheless, some organisms, such as fungi, can adapt to these extreme conditions. The environmental temperature is one of the parameters thoroughly affecting the structure and composition of fungal membrane lipids. The psychrophilic fungi generally increase the disorder within macromolecules to maintain membrane fluidity at low temperatures. To do so, Antarctic fungi increase the proportion of unsaturated fatty acids that allow maintaining a semi-fluid state of the membranes and survive at extremely low temperatures. This ecological feature may be of interest for the characterization of phenotypical traits of the ecological adaptation of these fungi to the extreme environmental conditions of Antarctica. Moreover, this can be of inspiration to find solutions inspired by nature for alternative sources of polyunsaturated fatty acids (PUFAs) for diets of humans and animals. We characterized three fungal strains isolated from Antarctica and set up a laboratory/small-scale production of fungal biomass with a high content of beneficial PUFAs. In detail, three fungal species previously isolated from Antarctic environmental matrices were tested and identified at the genome level. We also conducted growth experiments to determine the effects of temperature and substrate on biomass and PUFA production. The results showed that these fungi have a high percentage of PUFAs compared to saturated ones; the growth at low temperatures (10°C) increases the production of linolenic acid (C18:3) while the biomass amount (yield) depends on the composition of the growth substrate; a satisfying qualitative-quantitative production is achieved using agri-food chain waste products, such as brewing and whey waste, as a growth substrate.
|
|
Scooped by
mhryu@live.com
Today, 6:02 PM
|
Global climate change poses increasing threats to seed production and thus food security. The seed microbiome plays an essential role in regulating the whole seed life cycle. Specific seed endophytes and spermosphere microorganisms orchestrate the maintenance and termination of dormancy towards the synchronization of germination plasticity to meet agricultural demands. In this review, we summarize recent advances by linking seed-microbiome interactions with seed processes. We review the sources of seed microbiomes and their physiological regulation on dormancy and germination in response to environmental changes with a focus on phytohormone crosstalk. We also discuss the molecular mechanisms by which seed-microbe interactions affect seed destiny. Finally, we explore emerging precision applications of microbiomes in the seed industry by integrating cutting-edge technologies such as microbial seed coatings and artificial intelligence (AI) in seed science and technology. In conclusion, harnessing microbiome-based strategies to manipulate seed life cycle holds immense promise for sustainable food production in a changing global climate.
|
|
Scooped by
mhryu@live.com
Today, 4:21 PM
|
Advances in synthetic biology continue to potentiate bacterial cancer therapy. Here, we constructed biosensor-driven encapsulation systems for autonomous control of capsular polysaccharides of Escherichia coli Nissle 1917 to improve pharmacokinetic profiles. The engineered bacteria were programmed to express capsular polysaccharides for immune evasion upon intravenous administration to reach tumors and then turn off gene expression upon colonizing the tumors based on quorum-sensing or acid-sensing to prevent dissemination of bacteria into the systemic circulation. Because a classical pharmacokinetic model could not capture the dynamic nature of living therapeutics, a two-state pharmacokinetic model was developed to simulate the autonomous control of capsular polysaccharides in different biological compartments and their impact on biodistribution. Using this model, we identified parameters in gene circuit dynamics and immune clearance that influence tumor colonization and systemic bacterial persistence. In a “humanized” pharmacokinetic model with an increased rate of complement-mediated lysis of bacteria, biosensor-driven systems achieved tumor seeding densities comparable to wild-type bacteria while reducing bacterial loads in blood and liver by several orders of magnitude, highlighting their potential for safe systemic delivery. The biosensor-driven systems represent a more effective strategy to control living drugs than inducible systems, and the two-state pharmacokinetic model is a first step to capture the autonomous nature of this new class of therapeutics for clinical translation.
|
|
Scooped by
mhryu@live.com
Today, 3:56 PM
|
Gene expression is typically studied on a gene-by-gene basis, with regulation analyzed primarily in response to environmental or developmental cues. In contrast, much less is known about how intrinsic factors, such as cell size or DNA content, influence global gene expression patterns. Cell size varies significantly across different cell types and dynamically changes during the cell cycle. To maintain proper intracellular concentrations of biomolecules such as mRNAs and proteins, gene expression must be coordinated with cell size. Emerging evidence from diverse organisms, including bacteria, yeast, animals, and plants, demonstrates that transcriptional output scales with cell size, suggesting a conserved principle of gene regulation. However, the mechanisms by which cells sense their size and modulate gene expression accordingly remain poorly understood. In this review, we summarize recent advances in uncovering the molecular and cellular principles of gene expression scaling with cell size across kingdoms. We also highlight key open questions in the field, with a particular emphasis on how plant systems, still underexplored in this context, can provide additional insights into the fundamental principles of size-dependent gene regulation.
|
|
Scooped by
mhryu@live.com
Today, 3:41 PM
|
TnpB is a compact RNA-guided endonuclease and evolutionary ancestor of CRISPR-Cas12 that offers a promising platform for genome engineering. However, the genome-editing activity of TnpBs remains limited and its underlying determinants are poorly understood. Here, we used biochemical and single-molecule assays to examine the DNA-unwinding mechanism of Youngiibacter multivorans TnpB (Ymu1 TnpB). DNA unwinding proceeds through formation of a partially unwound intermediate state to a fully unwound open state. The open state forms inefficiently and collapses readily in the absence of negative supercoiling. An optimized variant, Ymu1-WFR, stabilizes formation of both the intermediate and open states, resulting in enhanced DNA cleavage in vitro and increased genome editing in vivo. These findings identify the physical basis for the observed minimal activities of natural TnpBs, revealing how stabilizing specific unwinding states enables efficient DNA targeting.
|
|
Scooped by
mhryu@live.com
Today, 3:15 PM
|
Generative protein modeling provides advanced tools for designing diverse protein sequences and structures. However, accurately modeling the conformational landscape and designing sequences remain critical challenges: ensuring that the designed sequence reliably folds into the target structure as its most stable conformation, and optimizing the sequence for a given suboptimal fixed input structure. In this study, we present a systematic analysis of jointly optimizing sequence-to-structure and structure-to-sequence mappings. This approach enables us to find optimal solutions for modeling the conformational landscape. We validate our approach with large-scale protein stability measurements, demonstrating that joint optimization is superior for designing stable proteins using a joint model (TrRosetta and TrMRF) and for achieving high accuracy in stability prediction when jointly modeling (half-masked ESMFold pLDDT + ESM2 Pseudo-likelihood). We further investigate features of sequences generated from the joint model and find that they exhibit higher frequencies of hydrophilic interactions, which may help maintain both secondary structure registry and pairing-features not captured by structure-to-sequence modeling alone. This study presents a comprehensive modelling framework that jointly optimizes sequence and structure to generate de novo proteins with improved folding stability, providing large-scale experimental benchmarking across multiple computational design methods
|
|
Scooped by
mhryu@live.com
Today, 2:22 PM
|
Translation is a fundamental process for every living organism. In plants, the rate of translation is tightly modulated during development and in responses to environmental cues. However, it is challenging to measure the actual translation state of the tissues in vivo. Here, we report the introduction of an in vivo translation marker based on bimolecular fluorescence complementation, the Ribo-BiFC. We combined a method originally developed for the fruitflies with an improved low background split-mVenus BiFC system previously described in plants. We labelled small subunit ribosomal proteins (RPS) and large subunit ribosomal proteins (RPL) of Arabidopsis thaliana with fragments of the mVenus fluorescent protein (FP). We tested the Ribo-BiFC method using transiently expressed recombinant ribosomal proteins in epidermal cells of Nicotiana benthamiana. The BiFC-tagged ribosomal proteins complemented the mVenus molecule and were detected by fluorescence microscopy, potentially visualizing the close proximity of translating assembled 80S ribosomal subunits. Although the resulting signal is less intense than that of known interactors, its detection points to the functionality of the system. This Ribo-BiFC approach has further potential for use in stable transgenic lines in enabling the visualization of translational rate in plant tissues and changing translation dynamics during plant development, under abiotic stress or in different genetic backgrounds.
|
|
Scooped by
mhryu@live.com
Today, 1:12 PM
|
Nutrient crossfeeding critically governs microbiome–host interactions and ecosystem stability. Cobamides, synthesized only by prokaryotes, offer a powerful and tractable model for studying nutrient-mediated interdependencies in soil food webs; however, their ecological role in sustaining soil health remains unclear. Here, we construct the Soil Cobamide Producer database (SCP v.1.0) by integrating over 48,000 metagenomic and genomic datasets from 1,123 sampling sites. This database catalogs phylogenetically diverse prokaryotes (19 phyla, 302 genera) with cobamide biosynthetic potential. Using this resource, we identify host-specific colonization patterns of cobamide-producing microbes in fauna. These microbes also carry diverse functional traits that may contribute to trophic cascades and microbial community stability. In an Enchytraeid model, these colonizers support host development, modulate gene expression, and promote gut stability through transkingdom interactions, with cobamide biosynthesis serving as one representative trait among multiple microbial functions. At macroecological scales, cobamide-producing microbes occur across relatively high trophic levels, reflecting a broader principle of nutrient transfer that may also apply to other essential metabolites. This framework provides a general basis for studying nutritional microbes in soil food webs and advances One Health research. Soil life relies on microbial nutrient exchange to maintain ecosystem balance. This study shows that cobamide-producing microbes act as essential connectors in soil food webs, supporting host health and highlighting cross-kingdom links central to One Health.
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
High-throughput sequencing and computational protein design have created a growing gap between the discovery of new proteins and their functional characterization. In many instances, functional characterization requires one-to-one measurements—such as when detailed biochemical insights are desired or pooled selections are not possible—necessitating that individual variants be isolated and assayed. A major barrier to closing this gap is the cost to directly synthesize individual genes, which remains prohibitively expensive ($10–100 per sequence) and restricts these studies to small subsets of relevant variants, leaving many sequences without functional annotation. To address this, we developed user-defined Sorted Mutants (uSort-M), which combines pooled DNA synthesis, automated cell sorting of transformed Escherichia coli, and long-read sequencing to rapidly isolate and identify variants from diverse libraries. uSort-M can isolate, sequence, and validate individual variants from pooled libraries produced via diverse existing methods including multiplex assembly, error-prone PCR, or pooled QuickChange mutagenesis. Sorting single bacterial clones into 384-well plates is efficient: eight plates (3,072 wells) can be filled in 1–2 hours, with up to 90% of wells yielding monoclonal cultures. Commercial long-read sequencing enables accessible, fast, and cost-effective identification of individual sequences from isolated clones while tolerating wide variation in fragment length and diversity across the library. Applying this workflow to a 328-member scanning mutagenesis library of a 300-bp gene recovered 96% of desired variants at fivefold lower cost than traditional synthesis. Numerical simulations identify key parameters governing library recovery and enable accurate prediction of the sampling effort required to achieve target coverage. As library size increases, this workflow offers substantial savings over traditional gene synthesis or cloning. Due to its generalizability, efficiency, and reliance on standard instrumentation, uSort-M removes a key barrier to large-scale protein functional characterization.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
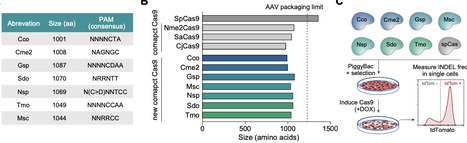
The development of compact and efficient CRISPR-Cas systems is crucial for biomedical and therapeutic genome editing, particularly in vivo applications based on viral delivery. Here, we performed a comparative functional screen of seven Cas9 orthologs to systematically evaluate their genome editing activity in mammalian cells. Among these, Cme2, a 1008 amino acid nuclease recognizing a 5' NAGNGC PAM, emerged as a promising candidate based on its compact size and baseline editing activity. To overcome its limited native efficiency, we employed a dual engineering approach combining sgRNA scaffold optimization and rational protein mutagenesis. The resulting variant, enCme2, exhibits markedly improved editing efficiency across multiple loci in both mouse and human cells while maintaining extremely high specificity and minimal off-target activity. Importantly, the small size of enCme2 permits packaging of the complete system into a single rAAV vector, enabling efficient genome editing/HDR in in vivo tissues and mouse embryos, and facile generation of transgenic models. These results establish enCme2 as a compact, precise, and AAV-compatible genome editing platform with broad applicability for in vivo research and therapeutic approaches, especially where high specificity is desirable.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
The rapid advancement of environmental sequencing technologies, such as metagenomics, has significantly enhanced our ability to study microbial communities. The eubiotic composition of these communities is crucial for maintaining ecological functions and host health. Species diversity is only one facet of a healthy community’s organization; together with abundance distributions and interaction structures, it shapes reproducible macroecological states, that is, joint statistical fingerprints that summarize whole-community behavior. Despite recent developments, a theoretical framework connecting empirical data with ecosystem modeling is still in its infancy, particularly in the context of disordered systems. Here, we present a novel framework that couples statistical physics tools for disordered systems with metagenomic data, explicitly linking diversity, interactions, and stability to define and compare these macroecological states. By employing the generalized Lotka–Volterra model with random interactions, we reveal two different emergent patterns of species interaction networks and species abundance distributions for healthy and diseased microbiomes. On the one hand, healthy microbiomes have similar community structures across individuals, characterized by strong species interactions and abundance diversity consistent with neutral stochastic fluctuations. On the other hand, diseased microbiomes show greater variability driven by deterministic factors, thus resulting in less ecologically stable and more divergent communities. Our findings suggest the potential of disordered system theory to characterize microbiomes and to capture the role of ecological interactions on stability and functioning.
|
|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
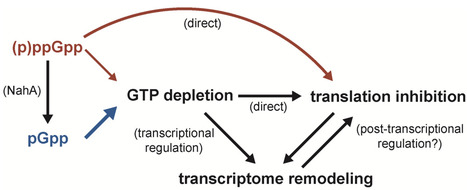
Bacteria produce the alarmone nucleotides (p)ppGpp during stress to affect replication, transcription, translation, and metabolism. Recently, pGpp was identified as a third alarmone that is produced from the hydrolysis of (p)ppGpp. Although pGpp is a major component of bacterial stress responses, its precise role in mediating these responses is poorly understood. ppGpp and pppGpp bind translation GTPases and therefore directly affect translation to conserve resources during periods of stress. Here, we show that while pGpp is a weaker inhibitor of protein synthesis than ppGpp and pppGpp in vitro, pGpp production in the model Gram-positive bacterium Bacillus subtilis leads to faster translation inhibition in vivo. Faster translation inhibition is accompanied by greater levels of disengaged ribosomal subunits and hibernating ribosome dimers, suggesting that translation initiation is strongly inhibited. We show that alarmone production in vivo causes a severe depletion of GTP, which is sufficient for translation inhibition. Finally, we find that pGpp production also causes more robust transcriptome remodeling than (p)ppGpp production. This work supports a model that implicates all three alarmones in translation inhibition via GTP depletion.
|