Your new post is loading...

|
Scooped by
mhryu@live.com
January 7, 11:54 PM
|
RNA secondary structures serve as a bridge between RNA sequences and often-unknown three-dimensional structures, offering insights into base pairing, structural motifs, and the overall organization of RNA molecules. To support efficient visualization and editing of these structures, we present Exornata, a modern, web-based editor designed to facilitate generation of detailed and standardized RNA secondary structure modeling. Exornata, is an expanded successor to the original XRNA software and is implemented using React and JavaScript/TypeScript technologies to ensure flexibility, interactivity, and high-quality rendering. Users can load, edit, and export RNA structures in multiple supported formats, ranging from conventional SVG to an advanced, in-house–developed JSON schema designed for interoperability with other resources such as R2DT, Traveler, and RiboVision2. The application supports multiple constraint-based editing modes (e.g. nucleotide, strand, helix, and domain) allowing precise and hierarchical manipulation of RNA elements. Exornata supports detailed, interactive visualization of canonical and non-canonical base pairs; it enables researchers to format and annotate structures, and to integrate them into broader bioinformatics pipelines. The tool is open-source, freely available at https://ldwlab.github.io/XRNA-React/, and is accompanied by a user guide.
|
|
Scooped by
mhryu@live.com
January 7, 11:11 PM
|
The ability to generate genomic diversity expands opportunities for understanding and engineering biology. Here, we demonstrate on-demand generation of diversity in bacterial genome configurations and its application to probing physiology under altered genome organization. We engineered the fast-growing bacterium Vibrio natriegens to enable large-scale stochastic duplications, translocations, inversions, and deletions, producing populations with a wide range of genome arrangements. We investigated phenotypic robustness to genome reconfigurations and found that distinct genome organizations can support stable physiology, indicating that bacteria may tolerate chromosomal alterations more readily than previously appreciated. Our work provides a framework for advancing the understanding and engineering of bacterial genomes and suggests that genome reconfigurations may contribute to phenogenetic drift, allowing for evolutionary exploration while preserving phenotype.
|
|
Scooped by
mhryu@live.com
January 7, 5:08 PM
|
Engineering DNA polymerases to efficiently synthesize artificial or noncognate nucleic acids remains an essential challenge in synthetic biology. Here we describe an evolutionary campaign designed to convert a family of highly selective DNA polymerases into an unnatural homolog with strong RNA synthesis activity. Starting from a homologous recombination library, a short evolutionary path was achieved using a single-cell droplet-based microfluidic selection strategy to produce C28, a newly engineered polymerase that can synthesize RNA with a rate of ~3 nt s−1 and of >99% fidelity. C28 is capable of long-range RNA synthesis, reverse transcription and chimeric DNA–RNA amplification using the PCR. Despite strong discrimination against other genetic systems, C28 readily accepts several 2′F and base-modified RNA analogs. Together, these findings highlight the power of directed evolution as an approach for reprogramming DNA polymerases with activities that could help drive future applications in biotechnology and medicine. Engineering polymerases to synthesize alternative genetic polymers remains a challenging problem in synthetic biology. Using DNA shuffling and droplet microfluidics, the current study provides a short evolutionary path from a DNA polymerase to one with robust RNA-synthesizing activity.
|
|
Scooped by
mhryu@live.com
January 7, 4:32 PM
|
Viable but non-culturable (VBNC) cells represent a reversible, metabolically active state that promotes the survival of bacteria under stressful conditions and their persistence in healthcare facilities and food industry. We conducted a systematic review following PRISMA 2020 guidelines to identify in vitro methodologies for inducing and resuscitating VBNC Enterobacteriaceae and Pseudomonas aeruginosa, and to determine key influencing factors. Eligible studies reported in vitro resuscitation of these species. Searches were performed in MEDLINE (PubMed), Scopus, and Google Scholar up to July 2025. Two independent reviewers screened and extracted data. Exclusion criteria included absence of original experimental data, focus on other species, or lack of clear VBNC definition. Risk of bias was qualitatively assessed. Analyses were descriptive without meta-analysis. Of the 1041 records, 24 articles (27 studies) were included. Resuscitation protocols typically employed standard culture media with additives and moderate incubation temperatures, with most successful recoveries occurring after 24–48 h. P. aeruginosa generally required less supplementation than Enterobacteriaceae. Reported mechanisms involved metabolic reactivation, oxidative stress modulation, nutrient sensing, and ribosome reactivation. The limitations of our study include protocol heterogeneity, lack of standardization, and selective reporting. While simple resuscitation methods were often effective, tailoring conditions to species-specific ecological preferences appears critical. Standardized approaches of VBNC cells will improve detection, risk assessment, and infection control.
|
|
Scooped by
mhryu@live.com
January 7, 4:24 PM
|
Site-specific DNA recombinases are powerful tools for genome engineering. The recent convergence of DNA recombinase-mediated technology with prime editing has established a promising new frontier for large-scale genomic integration. However, this strategy has limitations, including low intrinsic catalytic efficiency of DNA recombination enzymes and inefficiency of their long DNA landing pad insertions. To overcome these challenges, we developed a novel directed evolution strategy using progressively shortening landing pads as a selective pressure. The resulting evolved variants, VK and AVK, exhibited substantially enhanced intrinsic activity that is maintained on short-length landing pads. This enables a powerful synergistic effect with increased insertion efficiency of prime editing, leading to a dramatic increase in overall integration efficiency while maintaining high genomic specificity. This work thus provides a new class of highly efficient recombinases and a robust engineering strategy with broad applicability for both the development of gene therapies and fundamental research.
|
|
Scooped by
mhryu@live.com
January 7, 4:14 PM
|
As the number and complexity of RNA and DNA structures continue to expand, there is a growing need for robust yet accessible tools that support their accurate interpretation, validation, and refinement. We present DNATCO v5.0 https://dnatco.datmos.org/app an interactive web application for comprehensive structural analysis of nucleic acids. DNATCO integrates the NtC dinucleotide conformational classes and the CANA structural alphabet to provide an intuitive, geometrically complete description of local backbone and base orientations, complemented by interactive visualization of base pairing. The platform performs quantitative validation of conformational similarity and covalent bond lengths and angles, using newly established nucleic-acid valence-geometry standards. Quantitative validation encompasses the confal score and scattergrams mapping the fit between experimental electron density and geometry similarity to the closest NtC class. All outputs are downloadable. Integrated diagnostic tools help users identify unusual or problematic regions, explore alternative conformations, and generate torsion-restraint files for downstream. DNATCO v5.0 is implemented entirely client-side via WebAssembly, ensuring fast performance and preserving data privacy, and supports both PDB and user-provided structural models. By combining a rigorous geometric framework with an approachable interface, DNATCO enables both non-experts and specialists to evaluate nucleic-acid structures with greater confidence and to improve models in ways that support accurate biological interpretation.
|
|
Scooped by
mhryu@live.com
January 7, 4:02 PM
|
Noncoding single-nucleotide variants (SNVs) that alter transcription factor (TF) binding can affect gene expression and contribute to disease. Sequence-based methods can excel at predicting TF binding, but rely on training data and can exhibit TF-specific biases. Here, we propose a structure-guided approach for noncoding SNVs, using AlphaFold 3 (AF3) to model TF–DNA complexes and FoldX for downstream physics-based assessment. Benchmarked against single nucleotide polymorphism-systematic evolution of ligands by exponential enrichment (SNP-SELEX) data for six TFs (SPIB, ELK3, ETV4, SF-1, PAX5, and MEIS2), the FoldX-based strategy showed good agreement with experimental allele preferences. Interestingly, differences in AF3’s interface-predicted template modelling (ipTM) score aligned even more closely with SNP-SELEX results, generally surpassing energy-based metrics. Application to known disease-associated variants recapitulated most reported effects for TFs including NKX2-5, GATA3, and USF2A-USF1. In these examples, considering both ΔipTM and FoldX energies proved more reliable than either metric alone. While less accurate than state-of-the-art sequence-based methods, this work demonstrates that structural modelling can yield interpretable insights into how noncoding variants influence TF binding. By highlighting both the promise and limitations of AF3 in this context, our study provides a framework for complementary structural evaluation of regulatory variants.
|
|
Scooped by
mhryu@live.com
January 7, 3:55 PM
|
Some prokaryotes carry CRISPR-associated transposons (CASTs), Tn7-like elements that incorporate genes encoding CRISPR-Cas effectors. CAST insertion is directed by CRISPR-Cas effectors through RNA-guided DNA binding and interactions with transposition-associated proteins. Although efficient sequence-specific DNA integration requires both precise target DNA recognition and coordinated interactions between effectors and transposition-associated proteins, the underlying mechanism remains elusive. Here, we determined three cryo-EM structures of target DNA-bound type I-F3 TniQ-Cascade from Vibrio parahaemolyticus, revealing how Cas8/5 recognizes the protospacer adjacent motif (PAM) and identifying a key residue responsible for the cytidine preference at position -2 of the PAM. We revealed mismatch tolerance at the PAM-proximal site. Structural analyses showed that correct base pairing at the PAM-distal site correlates with conformational changes in the Cas8/5 helical bundle and TniQ, bending the DNA to guide its downstream region toward the transposition machinery. Together, these dynamic rearrangements at the PAM-distal region provide insights into the licensing mechanism of type I-F3 CAST transposition and highlight its potential for genome engineering applications.
|
|
Scooped by
mhryu@live.com
January 7, 3:49 PM
|
As the most important carbon source for bioproduction, glucose is transported and enters the central carbon metabolic pathway directly in most microorganisms. However, certain bacteria utilize membrane-bound dehydrogenases to oxidize glucose to gluconic acid (GA) in the periplasmic space. GA is subsequently converted either to 5-keto-d-gluconate by PQQ-dependent gluconate dehydrogenase (GADH) or to 2-keto-d-gluconate by FAD-dependent GADH, with the latter further oxidized to 2,5-diketo-d-gluconate. This review systematically examines the composition, distribution, physiological functions, and key enzymes of this oxidative pathway, alongside industrial applications of its metabolic products. Special emphasis is placed on metabolic engineering strategies─including bottleneck elimination, cofactor balancing, and chassis optimization─to overcome inherent regulatory constraints and enhance carbon flux toward target compounds. The potential of this pathway for the sustainable production of tartaric acid, 2,5-furandicarboxylic acid, vitamin C precursors, and phosphorus fertilizers is comprehensively assessed.
|
|
Scooped by
mhryu@live.com
January 7, 3:41 PM
|
Polyethylene terephthalate (PET) is one of the most widely used plastic materials, and its large-scale application has caused severe environmental pollution. Compared to traditional physical and chemical degradation methods, biological degradation is considered the most promising approach. In this context, this review starts with the current research status of PET plastic microbial degradation. Then, it summarizes the construction of strains for heterologous expression of PET-degrading enzymes, the development status of whole-cell catalysts, the innovative ideas of microbial consortia and microorganism-enzyme systems, and the application development of microorganism-functional material systems. In addition, this review includes the use of multiple characterization methods to monitor the degradation effects of PET and changes in strain characteristics, providing theoretical evidence for PET degradation research. Finally, the researches on valorization of PET degradation monomers through synthetic biology are discussed, underscoring the potential of microbial degradation in PET waste upcycling.
|
|
Scooped by
mhryu@live.com
January 7, 3:31 PM
|
In all domains of life, tRNAs mediate the transfer of genetic information from mRNAs to proteins. As their depletion suppresses translation and, consequently, viral replication, tRNAs represent long-standing and increasingly recognized targets of innate immunity. Here we report Cas12a3 effector nucleases from type V CRISPR–Cas adaptive immune systems in bacteria that preferentially cleave tRNAs after recognition of target RNA. Cas12a3 orthologues belong to one of two previously unreported nuclease clades that exhibit RNA-mediated cleavage of non-target RNA, and are distinct from all other known type V systems. Through cell-based and biochemical assays and direct RNA sequencing, we demonstrate that recognition of a complementary target RNA by the CRISPR RNA triggers Cas12a3 to cleave the conserved 5′-CCA-3′ tail of diverse tRNAs to drive growth arrest and antiphage defence. Cryogenic electron microscopy structures further revealed a distinct tRNA-loading domain that positions the tRNA tail in the RuvC active site of the nuclease. By designing synthetic reporters that mimic the tRNA acceptor stem and tail, we expanded the capacity of current CRISPR-based diagnostics for multiplexed RNA detection. Overall, these findings reveal widespread tRNA inactivation as a previously unrecognized CRISPR-based immune strategy that broadens the application space of the existing CRISPR toolbox. Cas12a3 nucleases constitute a distinct clade of type V CRISPR–Cas bacterial immune systems that preferentially cleave the 3′ tails of tRNAs after recognition of target RNA to induce growth arrest and block phage dissemination.
|
|
Scooped by
mhryu@live.com
January 7, 12:11 AM
|
Understanding the extrinsic and intrinsic environmental factors that influence lag phase duration is critical for developing strategies to control the growth of foodborne pathogens. Although the exponential growth rate can be predicted as a straightforward response to the growth environment, lag phase duration is difficult to predict because it depends on not only the current growth conditions but also the previous growth environment and physiological status of the bacterial cells. Therefore, this article aims to provide a comprehensive understanding of the dynamic, adaptable, and evolvable nature of the lag phase. It is based on relevant literature (experimental studies, literature summaries, observational data) on the effect of pre- and postgrowth environments. We discuss the modeling strategies employed to incorporate physiological heterogeneity and dynamic food environments into predictive modeling frameworks. Overall, we summarize the empirical and mechanistic modeling strategies for quantifying lag phase duration.
|
|
Scooped by
mhryu@live.com
January 6, 11:59 PM
|
New approaches to engineering plant genomes have the potential to improve agriculture. However, transgenes insertion and tissue culture have become bottlenecks to genome-editing technology becoming widely adopted and achieving the promise of targeted editing. Recent developments in particle bombardment and viral vector-mediated delivery can open doors to overcome these limitations.
|
|
|
Scooped by
mhryu@live.com
January 7, 11:50 PM
|
Aptamers are single-stranded oligonucleotides that bind molecular targets with high affinity and specificity, offering significant advantages over antibodies in therapeutic and diagnostic applications. However, their discovery through Systematic Evolution of Ligands by EXponential enrichment (SELEX) remains time-consuming, expensive, and susceptible to experimental biases. Here we present AptaBLE, a deep learning framework that combines pretrained protein and nucleic acid sequence encoders with a novel symmetric bidirectional cross-attention architecture to predict aptamer-protein binding interactions. This design enables robust generalization across diverse protein targets and ssDNA aptamer modalities while maintaining the ability to process variable-length sequences. We demonstrate that AptaBLE significantly outperforms existing methods, achieving superior accuracy in predicting aptamer-protein binding. Furthermore, we demonstrate two complementary de novo generation approaches that produce novel aptamers binding target proteins with desired specificity profiles and Kds as low as 31nM to-date. AptaBLE represents a significant advance in computational aptamer design, providing an accessible, sequence-based platform for accelerating therapeutic and diagnostic aptamer development.
|
|
Scooped by
mhryu@live.com
January 7, 11:07 PM
|
The ocean, Earth’s largest carbon reservoir, exerts a central role over atmospheric CO2 through its capacity to store carbon primarily as bicarbonate ions. Direct observations indicate that the global ocean has a net carbon uptake of 2.6–3.0 petagrams of carbon annually, representing nearly 30% of anthropogenic CO2 emissions. This review examines two principal domains of oceanic carbon cycling. The first concerns the natural uptake and storage of anthropogenic CO2, with emphasis on the response of the marine carbonate system and the spatial distribution of absorbed carbon. The second addresses emerging marine CO2 removal strategies, especially ocean alkalinity enhancement and macroalgae-based approaches. Ocean alkalinity enhancement aims to increase seawater buffering capacity to facilitate greater CO2 uptake, whereas macroalgae-based strategies rely on photosynthetic fixation and the subsequent storage of organic and inorganic carbon in various reservoirs. Effective implementation of these approaches necessitates rigorous monitoring, reporting, and verification frameworks to ensure their quantifiable efficacy and environmental integrity.
|
|
Scooped by
mhryu@live.com
January 7, 4:36 PM
|
Although genome sequencing technologies have advanced rapidly, microbial genomes still contain numerous genes with unknown functions, posing ongoing challenges for comprehensive genome annotation. Traditional annotation methods are constrained by a lack of scalable experimental techniques and the limitations of conventional homology-based computational approaches. Recent computational innovations, particularly deep learning, have substantially improved gene function prediction, facilitating more efficient annotation of transcription factors, enzymes and other protein classes. Integrating computational and experimental approaches has enabled the development of workflows that systematize gene function discovery, paving the way for faster, more accurate and comprehensive genome annotation. Continued refinement of these integrated methods holds great promise for deepening our understanding of microorganisms. Here we review recent advances in artificial intelligence for gene function discovery and discuss future directions for achieving interpretable and high-throughput artificial intelligence-guided annotation. In this Review, the authors discuss how artificial intelligence can aid microbial gene function discovery.
|
|
Scooped by
mhryu@live.com
January 7, 4:29 PM
|
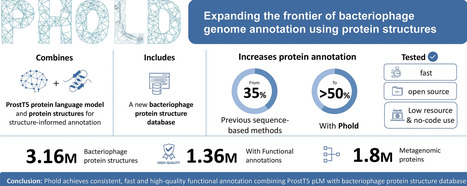
Bacteriophage (phage) genome annotation is essential for understanding their functional potential and suitability for use as therapeutic agents. Here, we introduce Phold, an annotation framework utilizing protein structural information that combines the ProstT5 protein language model and structural alignment tool Foldseek. Phold assigns annotations using a database of over 1.36 million predicted phage protein structures with high-quality functional labels. Benchmarking reveals that Phold outperforms existing sequence-based homology approaches in functional annotation sensitivity whilst maintaining speed, consistency, and scalability. Applying Phold to diverse cultured and metagenomic phage genomes shows it consistently annotates over 50% of genes on an average phage and 40% on an average archaeal virus. Comparisons of phage protein structures to other protein structures across the tree of life reveal that phage proteins commonly have structural homology to proteins shared across the tree of life, particularly those that have nucleic acid metabolism and enzymatic functions. Phold is available as free and open-source software at https://github.com/gbouras13/phold.
|
|
Scooped by
mhryu@live.com
January 7, 4:20 PM
|
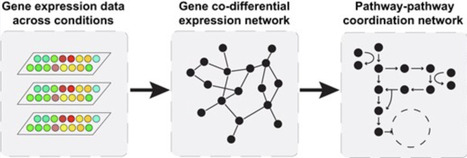
Gene co-expression networks are commonly used to identify functionally related genes, but they often suffer from spurious associations and fail to capture genes with restricted, context-specific responses. To address these limitations, we constructed YeastCoDEGNet, a global co-differential expression network in Saccharomyces cerevisiae, by reanalyzing microarray data from 143 carefully curated experiments comprising 425 comparisons. In this network, gene connections are defined by shared context-specific responses, enabling the identification of distinct topological groups enriched in either essential or nonessential genes, often associated with metabolic or nonmetabolic biological processes, respectively. We further characterized each gene by its responsiveness, essentiality, and number of co-differentially expressed partners, uncovering positional clustering of these features across chromosomal locations. To explore higher-order functional organization, we built a cross-pathway coordination network based on context-specific responses between pathway pairs, revealing modules of functionally related pathways. This network not only recovered well-known associations but also identified novel links, with particularly strong coordination observed among pathways involved in central carbon metabolism, amino acid and antioxidant processes, protein synthesis, trafficking, degradation, and gene regulation. By capturing context-specific gene expression dynamics, YeastCoDEGNet provides a powerful framework for studying how genes and pathways adapt to genetic and environmental perturbations.
|
|
Scooped by
mhryu@live.com
January 7, 4:08 PM
|
Magnetotactic bacteria produce membrane-bound organelles known as magnetosomes, which consist of chains of magnetite crystals and function as sensors for orientation within the Earth’s magnetic field. Magnetosome biosynthesis is a complex, multistep process that depends on iron availability and suboxic conditions. However, the expression of the ca. 30 core magnetosome biosynthetic genes has previously been described as constitutive and largely unaffected by environmental conditions. A recent study by Pang et al., published in this journal, reported the identification of a transcriptional regulator, NsrRMg, which was proposed to activate magnetosome biosynthesis in Magnetospirillum gryphiswaldense inresponse to the endogenous signaling molecule nitric oxide (NO). Furthermore, the study suggested that NO also regulates a putative, previously unrecognized nitrification pathway that presumably supports endogenous NO production via denitrification, even in the absence of extracellular nitrate. Here, we present results of genetic and transcriptional analyses demonstrating that, contrary to the findings of Pang et al., NsrRMg is not required for magnetosome biosynthesis. We also refute the existence of the proposed nitrification pathway and conclude that there is no compelling evidence supporting a role of NsrRMg as a key regulator of magnetosome formation in M. gryphiswaldense.
|
|
Scooped by
mhryu@live.com
January 7, 3:58 PM
|
RNA editing enzymatically modifies RNA molecules post-transcriptionally, enabling precise sequence alterations. Advantages include reversibility and temporal control without genomic DNA changes, allowing dynamic regulation of gene expression while preserving original genetic information. In this study, we characterized McAgo derived from Monosporascus cannonballus, which functions as a programmable nuclease guided by 14–30 nt gRNAs, demonstrating robust RNA cleavage activity at physiological temperature. Furthermore, we delivered McAgo RNP (ribonucleoprotein) complexes into mammalian cells, achieving >90% RNA knockdown efficiency with minimal innate immune responses. A catalytically inactive mutant (dMcAgo) using a gRNA as short as 20 nt, conjugated to the hADAR2 deaminase domain (hADAR2dd E488Q), achieved up to 90% RNA editing efficiency in vitro. This study establishes, for the first time, the effective targeting of endogenous RNA by a heterologous Argonaute in mammalian cells, alongside its demonstrated utility for RNA editing—thereby expanding the functional repertoire of Argonaute proteins.
|
|
Scooped by
mhryu@live.com
January 7, 3:51 PM
|
Soil salinization has been considered as a global problem in agriculture, which decreases crop productivity and threatens food security. Salt stress causes complex physiological damages in plants such as ionic imbalance, osmotic stress, and oxidative damage. However, plants have developed several genomic mechanisms to reduce these negative influences that are further supported by dynamic interactions with rhizosphere microbial communities. This review integrates current advances in understanding the interplay between plant genomes and the rhizosphere microbiome under salt stress. It highlights the role of plant-growth-promoting rhizobacteria (PGPR), arbuscular mycorrhizal fungi (AMF), and microbial volatiles in modulating gene expression and root architecture. Notably, PGPR such as Enterobacter sp. SA187 and Bacillus velezensis have been shown to upregulate key stress-related genes and increase antioxidant enzyme activities, which boost plant resilience under salinity. These microbes also influence stress signaling pathways such as SOS and ABA. Furthermore, this review also discusses the effect of root exudates on microbial communities, the application of synthetic microbial consortia, and genome-scale strategies such as transcriptomics, GWAS, and CRISPR. Our findings show that root exudation patterns shift significantly under salt stress, which enriches beneficial microbial taxa such as Sphingomonas and Streptomyces, while volatile compounds like benzenoids and ketones contribute to systemic stress responses. Understanding the synergistic plant–microbe interactions provides a foundation to engineer salt-resilient crops and for the advancement of sustainable agricultural practices in saline soils.
|
|
Scooped by
mhryu@live.com
January 7, 3:43 PM
|
Human breast milk (HBM) is composed of various components that are crucial for providing essential nutrients and enhancing infant immune system. Recently, the synthesis of HBM components through microbial fermentation has garnered significant attention due to its potential to reduce production costs, simplify manufacturing processes, and mitigate environmental pollution. However, this approach results in low yield and is difficult to scale up at the industrial level. Therefore, various synthetic biology tools have been used to enhance the efficiency of HBM component synthesis. This review first summarizes several synthetic biology tools that may improve HBM component production. Next, we have summarized HBM component production using microbial cell factories. Finally, we have summarized the challenges and opportunities presented by the construction of cell factories for the synthesis of HBM using synthetic biology tools. This article therefore provides a general guide to the construction of microbial cell factories for HBM components.
|
|
Scooped by
mhryu@live.com
January 7, 3:37 PM
|
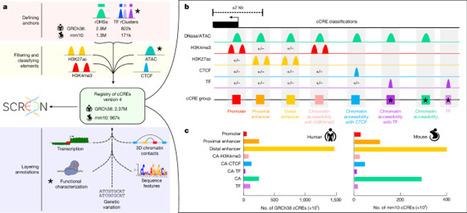
Mammalian genomes contain millions of regulatory elements that control the complex patterns of gene expression. Previously, the ENCODE consortium mapped biochemical signals across hundreds of cell types and tissues and integrated these data to develop a registry containing 0.9 million human and 300,000 mouse candidate cis-regulatory elements (cCREs) annotated with potential functions. Here we have expanded the registry to include 2.37 million human and 967,000 mouse cCREs, leveraging new ENCODE datasets and enhanced computational methods. This expanded registry covers hundreds of unique cell and tissue types, providing a comprehensive understanding of gene regulation. Functional characterization data from assays such as STARR-seq, massively parallel reporter assay, CRISPR perturbation and transgenic mouse assays have profiled more than 90% of human cCREs, revealing complex regulatory functions. We identified thousands of novel silencer cCREs and demonstrated their dual enhancer and silencer roles in different cellular contexts. Integrating the registry with other ENCODE annotations facilitates genetic variation interpretation and trait-associated gene identification, exemplified by the identification of KLF1 as a novel causal gene for red blood cell traits. This expanded registry is a valuable resource for studying the regulatory genome and its impact on health and disease. The existing ENCODE registry of candidate human and mouse cis-regulatory elements is expanded with the addition of new ENCODE data, integrating new functional data as well as new cell and tissue types.
|
|
Scooped by
mhryu@live.com
January 7, 12:16 AM
|
Small molecule–regulated protein oligomerization provides a powerful mechanism for manipulating biological processes by controlling protein proximity with high temporal precision. However, such systems only rarely exist in nature and remain a substantial challenge for de novo design. In this work, we describe a computational method for designing protein homooligomers whose assembly is regulated by small-molecule ligands with matching symmetry. We designed protein homotrimers regulated by the Food and Drug Administration (FDA)–approved drug amantadine and further designed amantadine-responsive heterodimers and heterotrimers. Biophysical characterization confirmed their amantadine-dependent assembly, and their crystal structures closely matched the design models. We demonstrated their broad applicability in controlling protein localization, membraneless condensate formation, and gene expression. Our approach opens new avenues for designing small molecule–responsive proteins and expands the chemogenetic toolkit for manipulating complex biological processes.
|
|
Scooped by
mhryu@live.com
January 7, 12:02 AM
|
Pseudomonas aeruginosa is a major opportunistic pathogen implicated in a wide range of infections, including chronic respiratory infections, burn wound infections, urinary tract infections, and device-associated infections. Its intrinsic and acquired resistance mechanisms, particularly its capacity for biofilm formation, pose serious challenges to conventional antibiotic therapy. With the continued rise of multidrug-resistant and pan-drug-resistant strains, the need for alternative therapeutic strategies has become increasingly urgent. Phages, viruses that specifically recognize and lyse bacteria, have shown unique advantages in combating antibiotic-resistant infections. This review systematically summarizes recent advances in the application of phage therapy for P. aeruginosa infections, covering in vitro bactericidal activity, biofilm degradation, and synergistic interactions with antibiotics. We further discuss evidence from animal models, including therapeutic efficacy, immunomodulatory effects, and pharmacokinetics. Emphasis is placed on clinical use cases, including different routes of administration, symptom relief, biomarker modulation, pathogen clearance rates, and adverse events. Typical case reports and early-phase clinical trials support the safety and efficacy of phage therapy. Nevertheless, translational barriers persist, such as the need for precise host matching, risks of immune neutralization, and the lack of standardized regulatory frameworks and Good Manufacturing Practice (GMP)-grade production systems. The rapid development of engineered phages and individualized therapeutic approaches offers a feasible path forward. In conclusion, phage therapy holds significant promise for the treatment of drug-resistant P. aeruginosa infections, and future efforts should focus on establishing standardized systems, conducting multicenter clinical studies, and leveraging synthetic biology to accelerate its translation from bench to bedside.
|