 Your new post is loading...

|
Scooped by
?
Today, 1:55 AM
|
Plant cell walls harbor vast carbohydrate reserves, yet how pathogens unlock them remains unclear. We show that the citrus canker pathogen Xanthomonas citri subsp. citri (Xcc) mobilizes cell wall sugars by hijacking a fruit-ripening program through the type III effector PthA4, which activates the ripening coordinator CsLOB1. CsLOB1 induces approximately 100 genes, many encoding enzymes involved in cell wall breakdown. In the nonfruiting species Nicotiana benthamiana, CsLOB1 likewise promotes Xanthomonas growth, showing that its activity is not strictly dependent on a ripening program. Transcriptomics and reporter assays revealed PthA4-dependent activation of the Xcc xylan CUT system, triggered by host-derived xylose and including a type II–secreted xylanase. Thus, PthA4-driven cell wall remodeling activates bacterial xylan use, establishing a TIII–TII effector feedforward loop that fuels Xcc proliferation.
|
|
Scooped by
?
Today, 1:35 AM
|
Enzymes are biological catalysts that speed up chemical reactions in an eco-friendly way. Precise enzyme design is hindered by vast sequence space and intricate sequence–structure–function interdependencies. To address these challenges, we developed EvoZymePro-Cat (EZPro-Cat), a deep learning platform for enzyme mutant screening. Conventional methods for predicting absolute mutant activities suffer from systematic errors and limited generalizability. Our pairwise comparison framework directly models relative activity superiority between variants, eliminating dependence on absolute value predictions. The framework integrates full sequence and local structure semantics of protein and ligand information using bilinear attention mechanisms. Protein sequences are encoded using the ESM1b transformer model. Ligands are represented through MolT5 embeddings and MACCS molecular fingerprints. The adaptability of protein residues to their microenvironments is captured by integrating structural features and site-specific evolutionary characteristics. Bilinear attention mechanisms capture long-range intermolecular interactions during catalysis by bidirectional projection and weighted fusion of protein–ligand features. Compared to existing methods, our model exhibits superior performance in identifying improved enzyme mutants through comparative prediction of mutation effects on activity, such as Km and kcat. For deep mutation scanning data sets, a few-shot learning strategy combined with the EZPro-Cat framework boosts prediction precision (AUC 0.908). By using integrated multimodal representations, EZPro-Cat offers a mechanistic and practical solution for functional profiling of intraprotein variants, driving paradigm shifts in highly efficient enzyme discovery and directed evolution.
|
|
Scooped by
?
Today, 1:25 AM
|
Arctic permafrosts are rapidly thawing in response to climate change, stimulating microbial activity and release of additional greenhouse gases, such as methane. Recently, catechin amendment of thawed permafrost soil microcosms demonstrated >80% decrease in methane production over 35 days compared to unamended controls, with metagenome, metatranscriptome and metabolome analyses revealing a shift in prokaryotic carbon metabolism from a syntrophic network feeding methanogens to one dominated by catechin fermentation. Here we leverage these same data to investigate potential virus impacts on this microbial community metabolism shift. In total, 900 DNA virus operational taxonomic units (vOTUs) were identified as actively lytic based on transcribed structural and lysis genes. Of these, 41% were predicted to infect at least one of 56 transcriptionally active prokaryote genera representing 13 phyla. The most active vOTUs were predicted to infect key catechin-degrading genera including Clostridium and undescribed Bacillota genus JAGFXR01. Temporally, viral communities responded later than prokaryotes to catechin amendment, reflecting a virus production lag to targeting key microbial responders. An induced prophage represented by a vOTU predicted to infect JAGFXR01 dominated the catechin-amended viral community response. It reached abundances 20-156 times higher than its host, suggesting intense viral lysis that could release degraded catechin intermediates. Indeed, catechin intermediate gene expression in non-catechin-degraders were elevated in catechin-amended samples, suggesting JAGFXR01 lysis products were taken up by non-catechin-degraders as part of community carbon metabolism rewiring. Together, these results potentially place viruses at the heart of carbon rewiring that modulates ecosystem outputs of climate-critical thawing permafrosts.
|
|
Scooped by
?
Today, 1:18 AM
|
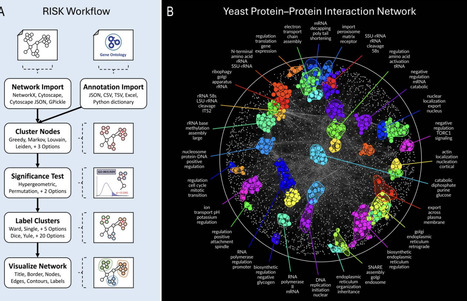
Analyzing biological networks demands scalable annotation tools, yet existing methods fall short in clustering power, statistical flexibility, and broad data compatibility. We introduce RISK (Regional Inference of Significant Kinships), a next-generation tool that overcomes these challenges by integrating community detection algorithms, rigorous overrepresentation analysis, and a modular architecture that supports diverse network types. RISK identifies biologically coherent relationships within networks and generates publication-ready visualizations, as demonstrated by its ability to resolve compact functional modules in Saccharomyces cerevisiae protein–protein interaction and genetic interaction networks. Its application to a high-energy physics citation network reveals structured relationships among research subfields, highlighting its versatility beyond biological systems. As biological and interdisciplinary networks increase in size and complexity, RISK’s scalability and adaptability make it a powerful solution for modern network analysis.
|
|
Scooped by
?
Today, 1:13 AM
|
Marine environments are frequently oligotrophic, characterized by low amount of bioassimilable nitrogen sources. At the global scale, the microbial fixation of N₂, or diazotrophy, represents the primary source of fixed nitrogen in pelagic marine ecosystems, playing a key role in supporting primary production and driving the export of organic matter to the deep ocean. However, given the high energetic cost of N₂ fixation, the active release of fixed nitrogen by diazotrophs appears counterintuitive, suggesting the existence of alternative passive release pathways that remain understudied to date. Here, we show that the marine non-cyanobacterial diazotroph Vibrio diazotrophicus is endowed with a prophage belonging to the Myoviridae family, whose expression is induced under anoxic and biofilm-forming conditions. We demonstrate that this prophage can spontaneously excise from the genome of its host and that it forms intact and infective phage particles. Moreover, phage-mediated host cell lysis leads to increased biofilm production compared with a prophage-free derivative mutant and to increased release of dissolved organic carbon and ammonium. Altogether, the results suggest that viruses may play a previously unrecognized role in oceanic ecosystem dynamics by structuring microhabitats suitable for diazotrophy and by contributing to the recycling of (in)organic matter.
|
|
Scooped by
?
Today, 12:47 AM
|
Pooled CRISPR screens using single-cell RNA sequencing (scRNA-seq) have emerged as powerful tools to uncover gene function, map regulatory networks, and identify genetic interactions. However, the inherent sparsity of bacterial scRNA-seq data has posed a major challenge toward applying these approaches to bacteria. Here, we present mapSPLiT, a pooled bacterial CRISPR activation and interference (CRISPRa/i) screening platform that enables large-scale experiments that simultaneously link hundreds of perturbations to their corresponding single-cell transcriptomes. By targeting 52 known or putative transcription factors with 118 perturbations in a pooled experiment, we expanded the E. coli regulatory network map, determined the function of putative regulators, and identified emergent global phenotypes. By targeting combinations of transcription factors simultaneously, we uncovered genetic interactions and regulatory logic between them. Mapping regulatory networks for carbon utilization in P. putida revealed control points that could expand metabolic flexibility and improve biomanufacturing. Together, these results establish mapSPLiT as a generalizable platform for single‑cell functional genomics in bacteria.
|
|
Scooped by
?
Today, 12:30 AM
|
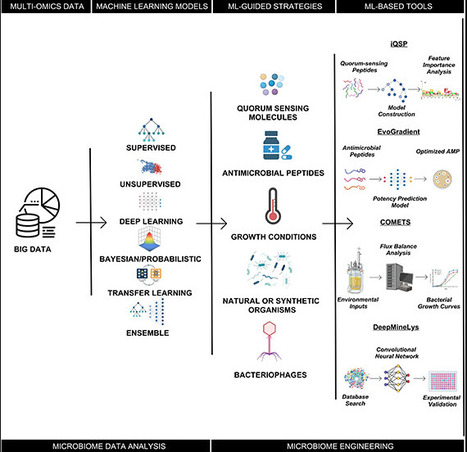
Microbiomes, complex communities of microorganisms and their genetic material, hold immense potential for addressing global challenges in diverse sectors, including healthcare, agriculture, and bioproduction. Engineering these intricate ecosystems, however, necessitates a comprehensive understanding of the complex web of microbial interactions. The emergence of machine learning (ML) has revolutionized microbiome research, offering powerful tools to analyze massive data sets, uncover hidden patterns, and predict microbial behavior. ML algorithms have demonstrated remarkable success in identifying and characterizing microbial communities, predicting interactions between organisms and optimizing the design of microbial communities for specific functions. This Perspective examines the transformative applications of ML in the context of microbiome engineering, encompassing both microbiome data analysis and the targeted manipulation of microbial communities. These techniques employ a variety of strategies, including the manipulation of quorum sensing molecules, antimicrobial peptides, growth conditions, the introduction of probiotics, and the utilization of bacteriophages. By integrating ML with experimental approaches, researchers are pushing the boundaries of microbiome engineering, paving the way for novel applications in diverse fields. However, it is important to acknowledge the challenges that ML algorithms face, such as the limited availability of high-quality, large-scale data sets, the inherent complexity of biological systems, and the need for improved integration of experimental and computational methods. This perspective further discusses the future perspectives of the field, highlighting expected developments in data generation, algorithm development, and interdisciplinary collaboration. These advancements hold the key to unlocking the full potential of microbial communities for addressing pressing global challenges.
|
|
Scooped by
?
Today, 12:22 AM
|
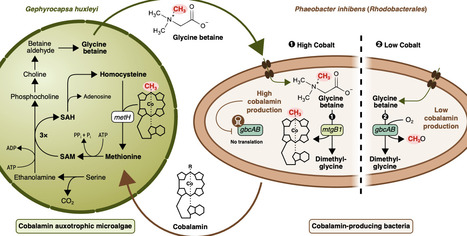
Heterotrophic bacteria supply cobalt-containing cobalamin (Cbl) to marine microalgae, which in return provide organic substrates. Such metabolic cross-feeding can regulate the species composition of prolific marine plankton and biofilms. Algal-produced glycine betaine (GB) can be catabolized by bacteria using a Cbl-dependent demethylase (MtgBCD). Yet, GB's impact on bacterial Cbl production during cross-feeding, and its control by cobalt scarcity remains elusive. Here, we demonstrate that Phaeobacter inhibens bacteria boost their Cbl production 25-fold when grown in monocultures with GB compared to glucose. During co-cultivation, P. inhibens satisfied the Cbl requirements of Gephyrocapsa huxleyi algae. Transcriptomic analysis of mono- and co-cultures revealed co-expression of mtgBCD and gbcAB genes encoding Cbl-dependent and -independent GB demethylases, respectively. Distinct growth defects of deletion mutants indicate that P. inhibens switches from Cbl-dependent to -independent GB demethylation under cobalt limitation, involving a Cbl riboswitch. Our findings suggest a positive feedback loop in which algal GB release stimulates the bacterial supply of Cbl. We predict the breakdown of this interaction under naturally occurring cobalt limitation which potentially contributes to the transient nature of algal blooms. Comparative genomics indicate that this mechanism is widespread in Rhodobacterales and other abundant marine Alphaproteobacteria, underscoring its pivotal role in global ocean productivity.
|
|
Scooped by
?
Today, 12:13 AM
|
Precise, reversible control of transgene expression is essential for safe and durable gene therapy, yet current inducible systems remain difficult to translate in vivo due to large size, limited induction duration, and dependence on immunogenic regulators. Here we present the adaptamer (ADAR modulatable aptamer), a compact (<120 bp) RNA switch that couples an FDA approved small molecule responsive aptamer with endogenous ADAR mediated RNA editing to regulate expression. Ligand binding stabilizes a double stranded RNA structure that recruits ADAR to convert a stop codon into a sense codon, restoring downstream protein translation. This cleavage free, post transcriptional mechanism enables precise, small molecule dependent modulation without exogenous protein machinery. We demonstrate that adaptamers are highly functional across multiple cell lines, including human T cells. In mice, AAV delivered adaptamer controlled FGF21 expression induced metabolic remodeling, significantly increasing energy expenditure and reversing obesity. This minimal, programmable system offers a clinically compatible approach for tunable genetic medicines and safe deployment of pleiotropic and dose limited proteins.
|
|
Scooped by
?
December 22, 11:58 PM
|
Type I toxin-antitoxin (TA) systems are widely distributed in the prokaryotic world, known for their antitoxin RNA interfering with the cognate toxin mRNA translation. Beyond their canonical role in plasmid maintenance, these systems represent evolutionarily optimized regulatory modules with rapid kinetics and modular architectures. Here, we repurposed them as post-transcriptional regulatory RNA devices via reverse engineering. After isolating the core of type I TA pairs and demonstrating the independence between their structure and repression function, we reconstructed artificial TA pairs termed SRTS-OPRTS. A platform for generating orthogonal SRTS-OPRTS pairs with cross-species application (Bacillus subtilis, Escherichia coli, and Corynebacterium glutamicum) was developed by introducing structure and energy constraints. As an individual expressing element or a co-expressing 3′ UTR tag within specific mRNA, SRTS achieved quantitative regulation of the gene with 3′ UTR cognate OPRTS. Such portability enabled convenient construction of dynamic mutually inhibitory switches, where genes tagged by SRTS and OPRTS could regulate each other. Leveraging this approach, a selective lethal system was further constructed to enrich high-fluorescent mutants, resulting in up to 11.32-fold enhancement in mean fluorescence intensity. Overall, these synthetic RNA devices provide portable tools for gene regulation and offer a robust foundation for constructing dynamic genetic circuits.
|
|
Scooped by
?
December 22, 11:43 PM
|
Antimicrobial resistance (AMR) is one of the most pressing global health challenges, often referred to as a ‘silent pandemic.’ Towards this, ARKbase is developed as an integrated, curated, value-added knowledgebase for AMR, with focus on WHO Bacterial Priority Pathogens. ARKbase has three core modules namely—Database, Insight, and Comparative Analysis Module—each offering curated data, including data analysed with custom-built pipelines. The Database Module has curated reference genomes and pan genomes along with antimicrobial susceptibility testing (AST) profiles. The Insight Module has 14 sub-modules and offers deep annotations encompassing antimicrobial resistance genes, virulence factors, structure annotation, host–pathogen interactions, biosynthetic gene clusters, known antibiotics, protein–protein interactions, drug targets, co-targets, drug-target interactions, machine learning models, and curated transcriptomic datasets for antibiotic exposure. The Comparative Analysis module offers a simple interface for comparing antimicrobial resistance genes, virulence factors, and drug targets among different priority pathogens. The data included in ARKbase are quality-checked and curated as per CLSI standards for AST, EUCAST guidelines for genome sequence, and the FAIR data principles. ARKbase is the first comprehensive knowledgebase focusing on WHO priority pathogens and AWaRe classification, enabling a combined evidence approach towards a holistic understanding of AMR. Available at https://datascience.imtech.res.in/anshu/arkbase.
|
|
Scooped by
?
December 22, 10:53 PM
|
Nanopore sequencing enables direct, single-molecule interrogation of biopolymers and shows promise for analyzing not only DNA and RNA but also chemically modified bases, proteins, and other polymers. Expanded DNA alphabets, such as those found in xenonucleic acids (XNAs), open new possibilities for diagnostics, therapeutics, data storage, and engineered biology. However, robust sequencing strategies for these modified molecules remain lacking. While nanopore-based tools exist for some noncanonical bases, they often require extensive experimental calibration by measuring each base across many sequence contexts, which limits scalability and increases cost. In this work, we investigate computational methods for predicting the ionic current signals produced during nanopore sequencing of DNA containing noncanonical XNA bases, aiming to reduce the need for experimental calibration. We compare a sequence-based predictive model with two structure-aware approaches: one using graph-based molecular representations and another adapting a generative language model to molecular SMILES. Our findings show that while sequence context captures much of the signal variability, incorporating structural and chemical information improves predictive accuracy in specific cases. These results highlight the value of structural data representations and model design in scaling XNA sequencing, and suggest this framework could extend to modeling ionic currents from other complex biomolecules, such as proteins.
|
|
Scooped by
?
December 22, 4:18 PM
|
De novo protein design is advancing rapidly, but many targets remain inaccessible to current AI-based tools. Here we describe de novo designed globular domains that drive biomolecular condensation. Starting from a water-soluble, monomeric protein, we make variants with the same amino-acid composition but different surface-charge distributions: one with large patches of surface charge, and another with a more-homogeneous charge distribution. The individual domains form stable and discrete structures in solution, with the large-patch variant exhibiting more-attractive interprotein interactions. Next, two copies of each variant are joined with disordered linkers to generate dumbbell-like proteins. When expressed in eukaryotic cells, the large-patch variant forms intracellular puncta, whereas that with small patches does not. The assemblies are dynamic, liquid condensates in vitro and in cells. The structured domains facilitate functionalisation: we introduce fluorophore-binding sites to visualize fluorescent condensates directly in cells without a GFP reporter; and we manipulate the condensates using motor proteins.
|
|
|
Scooped by
?
Today, 1:52 AM
|
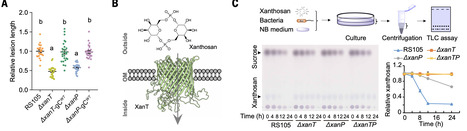
Xanthomonas spp. cause serious diseases in more than 400 plant species. The conserved AvrBs2 family effectors are among the most important virulence factors in xanthomonads, but how AvrBs2 promotes infection remains elusive. We found that AvrBs2 is a glycerophosphodiesterase-derived synthetase that catalyzes uridine 5′-diphosphate-α-d-galactose into a sugar phosphodiester, bis-(1,6)-cyclic dimeric α-d-galactose-phosphate, which is referred to as xanthosan. Xanthosan is synthesized by AvrBs2 in host cells and released into apoplastic spaces. Xanthomonas bacteria uptake xanthosan through the XanT transporter and hydrolyze it through the XanP phosphodiesterase for nutrition. AvrBs2, XanT, and XanP form a xanthosan “generation-uptake-utilization” system to provide a dedicated nutritional strategy to feed xanthomonads. Furthermore, elucidation of the AvrBs2-XanT-XanP virulence mechanism inspired us to develop an “anti-nutrition” strategy that should be applicable to control a wide variety of Xanthomonas diseases.
|
|
Scooped by
?
Today, 1:29 AM
|
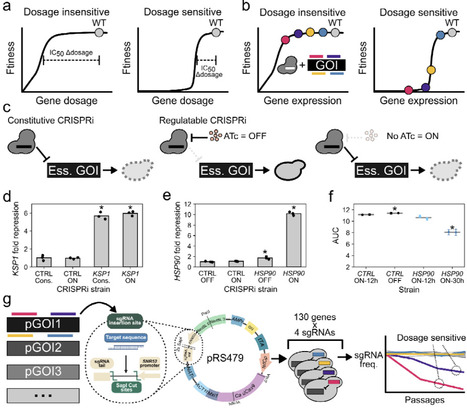
The rising rate of drug-resistant fungal infections and the emergence of fungal pathogens with intrinsic resistance phenotypes are a growing concern. The close evolutionary distance between mammals and fungi complicates the design of new antifungals and increases the chances of toxic off-target effects. As such, antifungal drug development usually focuses on fungal-specific proteins when considering potential new targets. Ideal drug targets should mediate essential cell processes and be highly sensitive to inhibition. Targeted gene repression can serve as a model for drug-mediated inhibition and for determining the dosage-sensitivity profile of genes of interest. In the fungal pathogen Candida albicans, classical approaches for gene repression can be labor-intensive and limited to one genetic background due to low throughput. Here, we adapt pooled CRISPRi in C. albicans for the first time and exploit this technique for large-scale functional genomic analysis. Through pooled CRISPRi screening, we test the repression sensitivity of over a hundred essential genes conserved in fungi but absent in humans in a single flask, and successfully identify highly dosage-sensitive genes across multiple cell components and pathways. We further extended our analysis to ten diverse environmental conditions, allowing us to measure how the environment influences dosage-sensitivity profiles. Finally, we extend our experiments to two clinical drug-resistant C. albicans strain backgrounds and demonstrate that many of the fitness defects we observed are conserved in resistant clinical isolates. Together, our results highlight a set of genes that are highly dosage-sensitive across different genetic and environmental contexts, making them attractive targets for further investigation. By facilitating rapid, efficient large-scale functional genomics assays across diverse genetic backgrounds, CRISPRi pooled screening will open new frontiers in C. albicans biology.
|
|
Scooped by
?
Today, 1:22 AM
|
Antibiotic persistence, characterized by a dormant subpopulation of bacterial cells that causes chronic and recurrent infections, remains poorly understood despite being recognized nearly a century ago. Toxin–antitoxin (TA) systems, which include a toxin and an antitoxin, are promising candidates for elucidating persister formation. We present the first theoretical model of persister formation driven by type I TA systems, in which the antitoxin is a small RNA molecule. Our analyses and simulations reveal two steady states—low toxin (normal growth) and high toxin (persistence)—with stochastic switching between them. Bistability requires both positive and negative feedback mediated by inhibition of antitoxin degradation. We derive stability diagrams that map mechanistic properties to system dynamics. The model suggests that while type I TA systems may not produce persisters under normal conditions, they can enter a bistable regime under stress, such as antibiotic exposure or nutrient limitation, leading to increased toxin expression or slower growth. Moreover, transiently slow-growing cells can be stabilized as long-living persisters through bistable TA dynamics. Using a cusp catastrophe surface, we identify distinct roles for two toxin inhibition mechanisms in modulating steady states and hysteresis. These findings provide a mechanistic basis for experimental observations and a framework for future studies.
|
|
Scooped by
?
Today, 1:15 AM
|
Metabolic network modeling is essential for understanding metabolic shifts occurring in complex physio-pathological processes. Currently, constraint-based modeling frameworks for metabolic networks primarily rely on Python or MATLAB libraries, requiring some coding skills. In contrast, more user-friendly tools lack essential features such as flux sampling or transcriptomic data integration We introduce COBRAxy, a Python-based tool suite integrated into the Galaxy Project. COBRAxy enables constraint-based modeling and sampling techniques, allowing users to compute metabolic flux distributions for multiple biological samples. The tool also enables the integration of medium composition information to refine flux predictions. Additionally, COBRAxy provides a user-friendly interface for visualizing significant flux differences between populations on an enriched metabolic map. This extension provides a comprehensive and accessible framework for advanced metabolic analysis, enabling researchers without extensive programming expertise to explore complex metabolic processes.
|
|
Scooped by
?
Today, 12:59 AM
|
Invertebrates, as the majority of macroscopic species on the Earth, are important resources for natural products. Chemical investigations of animals can date back to the early 20th century and have led to the discovery of thousands of compounds with diverse biological functions. These natural products can be structurally classified as terpenoids, polyketides, and alkaloids. Additionally, many compounds have been isolated from symbionts, leading to the widespread belief that animals lack the capability for secondary metabolism. Recent biochemical studies challenge this notion, revealing great potential for animal biosynthesis research. Animals possess larger genomes and more complex metabolic pathways, suggesting untapped biosynthetic potential. In contrast to microorganisms, studies on the biosynthesis of natural products in animals remain limited. Characterized genes represent only a small fraction of their vast genomes. The discovery of biosynthetic gene clusters suggests that the methods used to mine the biosynthetic genes of microorganisms may also be applicable to animals. The characterization of 4-vinylanisole in locusts demonstrates that the pathways lacking clear core biosynthesis enzymes still require multidisciplinary experimental approaches. In summary, further biosynthesis studies will expand methodological approaches and accelerate the characterization of remaining natural product pathways.
|
|
Scooped by
?
Today, 12:41 AM
|
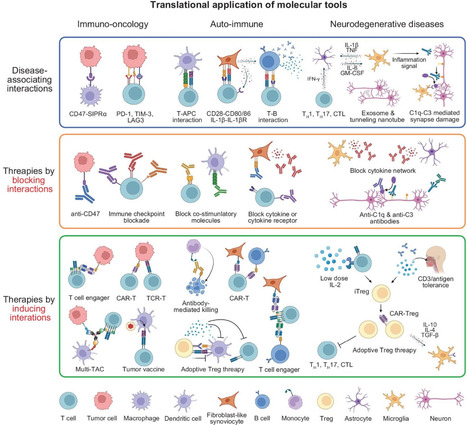
Cell-cell communication (CCC) is fundamental to essential biological processes including growth, differentiation, immune surveillance, and tissue homeostasis, and its dysregulation underlies various diseases such as cancer, autoimmunity, and neurodegeneration. In response to growing interest in decoding complex multicellular interactions, the 380th Shuangqing Forum entitled ‘Chemical, Biological, and Medical Frontiers in Multicellular Complex Systems’ was convened, providing a platform to discuss recent interdisciplinary breakthroughs. This review, emerging from forum discussions, highlights the latest advancements in molecular tools—such as super-resolution imaging, proximity labeling, bioorthogonal chemistry, synthetic receptors, and single-cell spatial omics—that enable unprecedented insights into spatial, molecular, and functional aspects of CCC. Emphasizing their translational potential, we discuss their profound implications for immuno-oncology, regenerative medicine, and autoimmune diseases. We further outline current challenges and opportunities, particularly advocating for a future precision medicine framework centered around targeted modulation of cell-cell interactions.
|
|
Scooped by
?
Today, 12:24 AM
|
Intra-strain variation in model bacterial pathogens can compromise experimental reproducibility and obscure biological interpretations. Dickeya solani, a necrotrophic potato pathogen, is widely studied using the type strain IPO 2222. However, phenotypic discrepancies among laboratories led us to investigate the genetic integrity of this reference stock. We identified at least three distinct IPO 2222 variants co-existing in the original stock, differing solely by mutations in the gene encoding the small regulatory RNA (sRNA) ArcZ. These findings resolve conflicting reports of antimicrobial activity in this strain described by Brual et al. (PLOS Genetics 19, e1010725, 2023) and Matilla et al. (mBio, e02472-22, 2022). We demonstrate that these are adaptive mutations rapidly selected in planta during host infection. Crucially, rather than systematically inactivating the gene, these mutations modulate the cellular levels of processed ArcZ. This modulation can uncouple virulence from antimicrobial activity. These variants behave as social cheaters, exhibiting a frequency-dependent fitness advantage over the cooperative wild-type strain during co-infection. These findings provide evidence that remodeling of a pleiotropic sRNA drives the emergence of bacterial cheaters within a plant host. The speed at which these mutants sweep through the population underscores the intense selective pressure acting on regulatory networks during infection, identifying sRNA modulation as a pivotal mechanism for rapid short-term adaptation.
|
|
Scooped by
?
Today, 12:18 AM
|
Microbial interactions shape the composition and stability of the human gut microbiome. Yet, the molecular mechanisms underlying these relationships remain poorly understood. Here, we systematically profiled 1,224 pairwise growth interactions among 36 representative gut bacterial strains using a spent medium assay. Most interactions were inhibitory and largely explained by resource depletion or medium acidification. We investigated further the molecular basis of a positive interaction, showing that Clostridium perfringens promotes the growth of Mediterraneibacter gnavus through extracellular vesicles. Additionally, we identified Veillonella parvula as a key species capable of modulating environmental pH, thereby enabling the growth of Parabacteroides merdae, a strain highly sensitive to acidic conditions. This pH-buffering effect was enhanced by guanine supplementation and persisted in multi-species communities containing different pH-lowering strains from diverse bacterial phyla. This demonstrates the ecological relevance of V. parvula, a species that, despite being commonly present in human gut microbiomes, is generally found at low levels. Overall, the comprehensive dataset and mechanistic insights presented here provide a foundation for rational strategies to manipulate microbiome composition and function.
|
|
Scooped by
?
Today, 12:07 AM
|
Plant genome editing has undergone a transformative shift with the advent of advanced molecular tools, offering unprecedented levels of precision, flexibility and efficiency in modifying genetic material. While classical site-directed nucleases such as ZFNs, TALENs and CRISPR-Cas9 have revolutionized genome engineering by enabling targeted mutagenesis and gene knockouts, the landscape is now rapidly evolving with the emergence of novel systems that go beyond the conventional double strand break (DSB)-mediated approaches. Advanced and recent tools include LEAPER, SATI, RESTORE, RESCUE, ARCUT, SPARDA, helicase-based approaches like HACE and Type IV-A CRISPR system, and transposon-based techniques like TATSI and piggyBac. These tools unlock previously inaccessible avenues of genome and transcriptome modulation. Some of these technologies allow DSB-free editing of DNA, precise base substitutions and RNA editing without altering the genomic DNA, a significant advancement for regulatory approval and for species with complex genomes or limited regeneration capacity. While LEAPER, RESCUE and RESTORE are the new advents in the RNA editing tool, SATI allows DSB-free approach for DNA editing, ARCUT offers less off-target and cleaner DNA repairs and Type IV-A CRISPR system induces gene silencing rather than editing. The transposon-based approaches include TATSI, piggyBac and TnpB, and helicases are used in HACE and Type IV-A CRISPR system. The prokaryotic Argonaute protein is used in SPARDA tool as an endonuclease to edit DNA. The transient and reversible nature of RNA editing tools such as RESTORE and LEAPER introduces a new layer of epigenetics-like control in plant systems, which could be harnessed for tissue-specific and environmentally-responsive trait expression. Simultaneously, innovations like ARCUT and SPARDA utilize chemically-guided editing, minimizing reliance on biological nucleases and reducing off-target risks. Their modularity and programmability are enabling gene function studies, synthetic pathway designs and targeted trait stacking. These advances represent a novel synthesis of genome engineering and systems biology, positioning plant genome editing not just as a tool of modification but as a platform for designing adaptive and intelligent crops, tailored to future environmental and nutritional challenges. Although, many of these recent tools remain to be applied on plant systems, they are proven to be effective elsewhere and hold a great potential to be effective in creating climate-resilient crops.
|
|
Scooped by
?
December 22, 11:46 PM
|
Protein structure prediction has a long history of benchmarking efforts such as critical assessment of structure prediction, continuous automated model evaluation and critical assessment of prediction of interactions. With the rise of artificial intelligence-based methods for prediction of macromolecular complexes, benchmarking with large datasets and robust, unsupervised scores to compare predictions against a reference has become essential. Also, the increasing size and complexity of experimentally determined reference structures by crystallography or cryogenic electron microscopy poses challenges for structure comparison methods. Here we review the current state of the art in scoring methodologies, identify existing limitations and present more suitable approaches for scoring of tertiary and quaternary structures, protein–protein interfaces and protein–ligand complexes. Our methods are designed to scale efficiently, enabling the assessment of large, complex systems. All developments are available in the structure benchmarking framework of OpenStructure. OpenStructure is open source software and available for free at https://openstructure.org/ . This study advances methods for benchmarking macromolecular complex predictions by introducing a scalable open-source framework used in recent community assessments to compare structures, interfaces and ligand interactions against experimental data.
|
|
Scooped by
?
December 22, 10:58 PM
|
CRISPR-cas provide sequence-specific mechanisms for targeting foreign DNA or RNA and have been widely used in genome editing and DNA detection. Type V CRISPR–Cas systems are characterized by a single RNA-guided RuvC domain-containing effector, Cas12. Here, through comprehensive mining of large-scale genomic and metagenomic data from microbial sources, we identified a new Class 2 CRISPR–Cas effector superfamily, designated Casδ, comprising three members with protein sizes ranging from 867 to 936 amino acids. Biochemical analyses revealed that Casδ-1 functions as a single RNA-guided endonuclease with specific recognition of 5′-RYR-3′ protospacer-adjacent motifs, where R represents A or G, and Y represents T or C. Casδ-1 exhibits robust double-stranded DNA cleavage activity and target-dependent trans-cleavage activity. Casδ-1 mediates efficient genome editing across species, achieving up to 60% indel rates in human cells while generating homozygous knockout lines in two agriculturally important monocot species (Oryza sativa and Zea mays) through stable transformation. Structural and evolutionary analyses reveal Casδ as an evolutionary transitional nuclease bridging Cas12n and canonical type V systems, featuring a C-terminal loop that is essential for activity. Collectively, Casδ is an evolutionarily distinct, compact (<1000 aa), tracrRNA-free CRISPR system enabling versatile cross-kingdom genome editing.
|
|
Scooped by
?
December 22, 9:55 PM
|
Transfer RNA (tRNA) modifications tune translation rates and codon optimality, thereby optimizing co-translational protein folding. However, the mechanisms by which tRNA modifications modulate codon optimality and trigger phenotypes remain unclear. Here, we show that ribosomes stall at specific modification-dependent codon pairs in wobble uridine modification (U34) mutants. This triggers ribosome collisions and a coordinated hierarchical response of cellular quality control pathways. High-resolution ribosome profiling reveals an unexpected functional diversity of U34 modifications during decoding. For instance, 5-carbamoylmethyluridine (ncm5U) exhibits distinct effects at the A and P sites. Importantly, ribosomes only slow down at a fraction of codons decoded by hypomodified tRNA, and the decoding speed of most codons remains unaffected. However, the translation speed of a codon largely depends on the identity of A- and P-site codons. Stalling at modification-dependent codon pairs induces ribosome collisions, triggering ribosome-associated quality control (RQC) and preventing protein aggregation by degrading aberrant nascent peptides and messenger RNAs. Inactivation of RQC stimulates the expression of molecular chaperones that remove protein aggregates. Our results demonstrate that loss of tRNA modifications primarily disrupts translation rates of suboptimal codon pairs, showing the coordinated regulation and adaptability of cellular surveillance systems. These systems ensure efficient and accurate protein synthesis and maintain protein homeostasis.
|




























r-1str