Your new post is loading...

|
Scooped by
?
Today, 3:54 PM
|
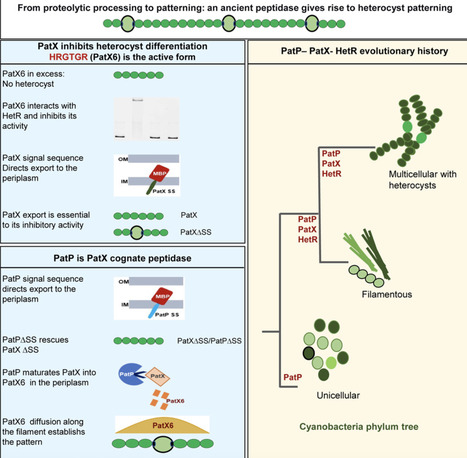
Spatial patterning in multicellular organisms is commonly explained by Turing-type reaction-diffusion systems, but the maturation of diffusible inhibitors remains poorly understood. In the cyanobacterium Nostoc PCC 7120, nitrogen deprivation triggers a pattern of nitrogen-fixing heterocysts regulated by HetR and inhibitory peptides, including PatX. We uncover the post-translational mechanism controlling PatX maturation, demonstrating its export and subsequent processing by the peptidase PatP. We identify HRGTGR, a PatX-derived hexapeptide, as the direct inhibitor of HetR, linking maturation to suppressed differentiation. Genomic analyses reveal that patP is ancient and conserved across all cyanobacteria, predating the patX-hetR module found only in filamentous clades. We therefore propose that this ancient peptidase was co-opted to process a new ligand, transforming a proteolytic event into a spatial patterning mechanism. This repurposing parallels eukaryotic signaling, underscoring a universal principle in the emergence of multicellular organization and providing a model for how complex patterns evolve from “simple” components.
|
|
Scooped by
?
Today, 3:40 PM
|
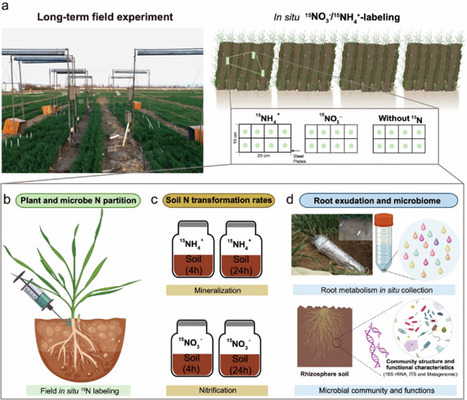
Sustainable crop production in a warming climate requires land management strategies that support plant-soil-microbe interactions to optimize nitrogen (N) availability. Here, we investigate the interacting effects of 10 years’ experimental warming and management (conservation vs. conventional agriculture) on wheat N acquisition using in situ 15N-labeling, root metabolomics and microbial metagenomics. We find that warming amplifies the positive effects on wheat nitrate uptake by 25% in conservation agriculture compared to conventional agriculture, while alleviating microbial competition for N. Additionally, warming increases soil gross N mineralization and nitrification rates by 191% and 159%, but decreases microbial immobilization by 24% in conservation agriculture. Concurrently, microbial genes for mineralization and nitrification are enriched, while those for N immobilization and nitrate reduction are reduced under conservation agriculture with warming. These shifts are driven by alterations in root primary and secondary metabolites, which reshape N-cycling microbial functional niches and optimize multiple microbial N processes beyond mere organic N mining. This reconfiguration increases carbon-nitrogen exchange efficiency, enabling wheat to outcompete soil microorganisms for N. Collectively, our findings suggest that conservation agriculture enhances plant N acquisition by strengthening plant-soil-microbe interactions under climate change, providing a sustainable strategy for future food security. Sustainable food production under climate change requires farming practices that support plant–soil–microbe interactions. This study suggests that conservation agriculture with warming enhances wheat nitrogen uptake by reducing microbial competition.
|
|
Scooped by
?
Today, 1:17 PM
|
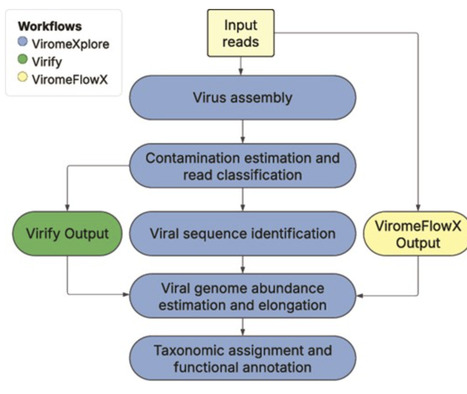
Viruses play a crucial role in shaping microbial communities and global biogeochemical cycles, yet their vast genetic diversity remains underexplored. Next-generation sequencing technologies allow untargeted profiling of metagenomes from viral communities (viromes). However, existing workflows often lack modularity, flexibility, and seamless integration with other microbiome analysis platforms. Here, we introduce “ViromeXplore,” a set of modular Nextflow workflows designed for efficient virome analysis. ViromeXplore incorporates state-of-the-art tools for contamination estimation, viral sequence identification, taxonomic assignment, functional annotation, and host prediction while optimizing computational resources. The workflows are containerized using Docker and Singularity, ensuring reproducibility and ease of deployment. Additionally, ViromeXplore offers optional integration with QIIME 2 and MOSHPIT, facilitating provenance tracking and interoperability with microbiome bioinformatics pipelines. By providing a scalable, user-friendly, and computationally efficient framework, ViromeXplore enhances viral metagenomic analysis and contributes to a deeper understanding of viral ecology. ViromeXplore is freely available at https://github.com/rhernandvel/ViromeXplore.
|
|
Scooped by
?
Today, 12:51 PM
|
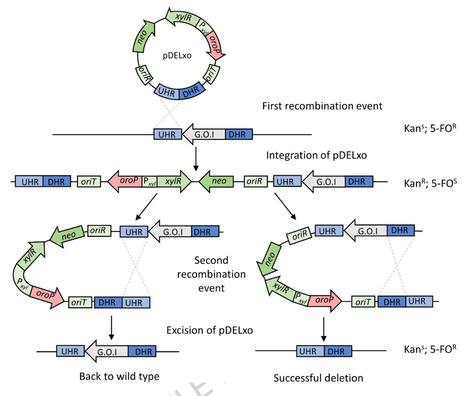
We here present the establishment of a counterselection system conducive to genome modifications in Bacillus methanolicus MGA3. We first identified four candidate genes or operons feasible to become counterselection markers: lacZ from Bacillus coagulans, sacB from Bacillus subtilis, codBA from E. coli, and oroP from Lactococcus lactis, based on their absence from the genome of B. methanolicus. We tested substrates of the encoded enzymes to confirm their lack of toxicity to wild type B. methanolicus. Experimental results confirmed that none of the tested substrates affected the growth of B. methanolicus wild type at physiologically relevant concentrations. Subsequently, the selected genes were individually cloned into a low-copy plasmid pTH1mp and used to transform B. methanolicus. We evaluated the conversion of these non-toxic substrates to toxic products upon heterologous expression of the respective marker genes in B. methanolicus. The recombinant strains were demonstrated to possess the desired counterselection activity through lack of growth in the presence of their relevant substrate. A novel transconjugation method for high-efficiency plasmid-delivery of B. methanolicus was developed and used for the establishment of genome modification via non-replicating suicide vector designed for homologous recombination. Deletion of the chromosomal upp gene, crucial for uracil metabolism, was achieved using this method. The deletion strain exhibited reduced sensitivity to 5-fluorouracil, the toxic substrate of the upp encoded enzyme, demonstrating the practical application of the counterselection markers in genome engineering of B. methanolicus.
|
|
Scooped by
?
Today, 12:03 PM
|
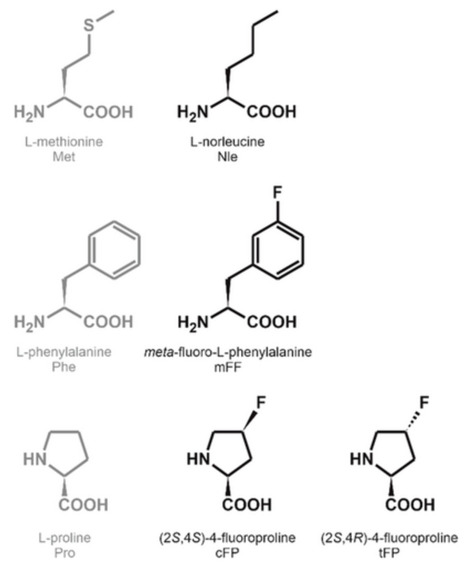
This study presents a robust bioprocess for the global incorporation of noncanonical amino acids (ncAAs) into proteins, enabling gram-scale production in auxotrophic Escherichia coli strains. The two-phase approach adapts from shake flask to bioreactor cultures and relies on cost-effective synthetic minimal media with glucose as the sole carbon source and yeast extract as an amino acid supply. It supports both external ncAA supplementation and in situ biosynthesis. A versatile E. coli BL21(DE3) auxotroph platform ensures broad ncAA and protein compatibility. Model proteins, such as a thermophilic lipase (TTL) and an oxidoreductase are labeled with biosynthesized norleucine (Nle), synthetic fluoroprolines, and fluorophenylalanine. Under optimal conditions, we achieved titers of up to 2 g L−1 with near-quantitative incorporation. To demonstrate the utility of the bioprocess for applications that require substantial amounts of proteins, the crystal structure of Nle-labeled TTL is solved. Future work should optimize media composition and feeding strategies to improve ncAA bioavailability and integrate biosynthesis pathways into the host genome to reduce metabolic burden and eliminate antibiotic use. These advances will make the process a cost-effective industrial platform for designer protein production.
|
|
Scooped by
?
Today, 11:02 AM
|
In biotechnological applications, it is often necessary to introduce genes or entire pathways into a host cell, which can create a significant metabolic burden on the host, limiting productivity. In this study, we systematically investigated the physiological stress responses of Pseudomonas putida during heterologous protein production using a modular monitoring system consisting of a plasmid encoding a heterologous protein fused to eGFP and a chromosomally integrated capacity reporter. Our findings reveal that translation is the main bottleneck, with translational capacity becoming saturated under high expression loads. While increasing the strength of the RBS improved protein production for non-burdensome proteins, this effect was not observed for larger fusion proteins. Variations in fusion protein size suggested that it is not the overall mass of the produced protein, but rather the length of the mRNA transcript, that contributes to metabolic burden. We further evaluated how resource availability affects protein expression by modifying the metabolic regime or supplementing with amino acids. While the carbon source affected cellular capacity, it did not significantly alter heterologous protein production. Amino acid supplementation alleviated the growth defects of MBPeGFP-producing cells and modestly improved protein production rates. Together, these findings emphasize that metabolic burden is influenced not only by the size of the produced protein but also by transcript architecture, resource allocation, and the physiological state of the host. Therefore, successful optimization of heterologous protein production requires a holistic approach integrating construct design with host physiology and cultivation strategies.
|
|
Scooped by
?
Today, 10:07 AM
|
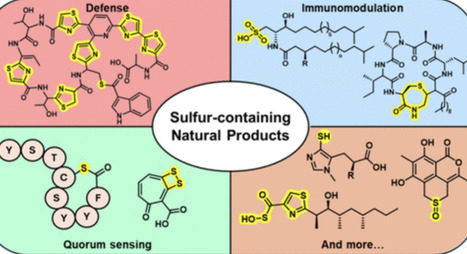
Natural products remain a primary source of new chemical entities and a central medium of biological communication. Among them, sulfur-containing natural products from microbes stand out for their diverse motifs─such as thioether (S–Cα/β/γ), thiazole, and sulfonate groups─distributed across RiPPs, NRPs, PKs, lipids, terpenoids, and hybrids. These sulfur-containing metabolites functionally contribute to communal interactions─intra- and interspecies microbe–microbe and microbe–host interactions─through their antibacterial, antifungal, antiviral, anticancer, and immunomodulatory activities. Crucially, they may act as information-rich signals that tune quorum circuits, biofilms, membrane and ion homeostasis, and host pathways. This review provides a structure- and mechanism-guided overview of sulfur-containing natural products reported over the past decade (2014–2025), covering their microbial sources, chemical diversity, as well as mechanisms of action and emphasizing how sulfur functionality encodes interaction strategies from host microbiomes to environmental systems.
|
|
Scooped by
?
Today, 9:41 AM
|
Mitochondria, which evolved from symbiotic bacteria, possess their own genomes (mtDNA) and support independent transcription and translation within the organelle. Given the essential role of mtDNA in energy production, metabolism, as well as cellular homeostasis, and the high density of confirmed pathogenic mutations that map to mtDNA, there is a pressing need for versatile methods to study and manipulate this genome. Although CRISPR technology has revolutionized the editing of nuclear genomes, it has not been successfully extended to mtDNA, primarily due to the challenge of delivering single guide RNAs (sgRNAs) across both outer and inner mitochondrial membranes. Here we develop a survival-based reporter in Saccharomyces cerevisiae to screen for potential RNA import motifs. We identify a 40-nucleotide aptamer (IM83) that facilitates sgRNA entry into the mitochondrial matrix, enabling CRISPR editing by a mitochondrially-localized adenine base editor. We show that mitochondrial import of IM83 is ATP-dependent and enhanced by the tRNA synthetase Msk1. Further investigations identify additional barriers to efficient CRISPR editing of mtDNA, including loss of membrane potential associated with mitochondrial targeting of the base editor. These insights lay the groundwork for future improvements in CRISPR-based editing of mtDNA in eukaryotes.
|
|
Scooped by
?
Today, 9:36 AM
|
Receptor signalling determines cellular responses and is crucial for defining specific biological outcomes. In legume root cells, highly similar and structurally conserved chitin and Nod factor receptor kinases activate immune or symbiotic pathways, respectively, when chitinous ligands are perceived. Here we show that specific amino acid residues in the intracellular part of the Nod factor receptor NFR1 control signalling specificity and enable the distinction of immune and symbiotic responses. Functional investigation of CERK6, NFR1 and receptor variants thereof revealed a conserved motif that we term Symbiosis Determinant 1 in the juxtamembrane region of the kinase domain, which is key for symbiotic signalling. We show that two residues in Symbiosis Determinant 1 are indispensable hallmarks of NFR1-type receptors and are sufficient to convert Lotus CERK6 and barley RLK4 kinase outputs to enable symbiotic signalling in Lotus japonicus. An investigation of plant receptor-like kinases identifies regions of these proteins that control whether immune or symbiotic signalling pathways are activated, with minimal changes to specific residues in one of these regions being sufficient to alter signalling specificity.
|
|
Scooped by
?
Today, 12:22 AM
|
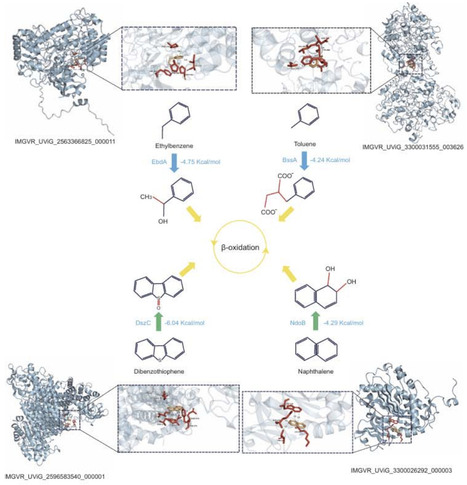
Hydrocarbons are vital for energy production and lead to serious environmental issues due to pollution. Microorganisms largely drive the production and degradation of hydrocarbons, yet little is known about viral contributions to hydrocarbon degradation. Here we identified 786 viral contigs from IMG/VR(v4), encoding five aerobic (alkB, ladA, almA, ndoB and dszC) and three anaerobic (bssA, ebdA and abcA) hydrocarbon-degrading genes (HDGs). vOTUs encoding HDGs span 26 distinct viral families, including 249 are mainly associated with host-associated environment and 463 are derived from aquatic habitat, respectively, implying that these viruses are broadly engaged in hydrocarbon-degradation processes across the entire biosphere. alkB (35%) and almA (28%) was tended to be encoded by Schizomimiviridae, bssA was tended to be encoded by T5-like bacteriophages (29%) and SPO1-like bacteriophages (22%), suggesting that the carriage of HDGs by viruses exhibits taxonomic specificity. With respect to host associations, Pseudomonadota and Bacillota constitute the two main potential host lineages, associated with 268 and 71 vOTUs, respectively. The profiling of ecological footprint reveals that viruses contribute a total of up to 30% gene abundance and 32% transcribing activity to hydrocarbon degradation in the global ocean. This study systematically revealed the distribution, diversity, virus-host correlation and activity of virus-encoded HDGs, underscoring the significant role of viruses in hydrocarbon metabolism on a global scale.
|
|
Scooped by
?
Today, 12:10 AM
|
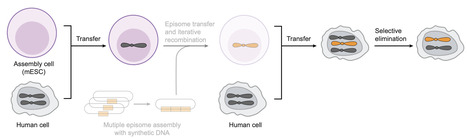
The synthesis of human genomes and other gigabase-scale genomes will require new strategies. Here, we realized key steps in our pipeline for building synthetic human chromosomes. We established: (i) the facile transfer of human chromosomes from human cells to mouse embryonic stem cells (assembly cells), where they are haploid, are nonessential, and may be operated on; (ii) the transfer of these human chromosomes from monochromosomal hybrids back into human cells to generate defined, synthetic aneuploidies; and (iii) the elimination of the corresponding endogenous human chromosomes to regenerate diploid cells containing a transferred chromosome. All steps were performed in nontransformed cells without chromothripsis and generated minimal structural variants, insertions, deletions, or single-nucleotide variants.
|
|
Scooped by
?
December 10, 11:44 PM
|
R-pyocins are phage tail-like protein complexes produced by Pseudomonas aeruginosa that deliver a single, lethal hit by depolarizing the target cell membrane. Unlike phages, R-pyocins lack capsids and DNA, and their killing is highly specific, being determined by tail fibre proteins that recognize subtype-specific LPS receptors on susceptible strains. Five known subtypes (R1–R5) vary in host range, with R5 displaying the broadest activity. R-pyocin expression is tightly regulated by the SOS response, linking their release to environmental stress. Their non-replicative mechanism and metabolic independence make them especially promising for targeting multidrug-resistant and biofilm-associated P. aeruginosa infections, such as those seen in cystic fibrosis and chronic wounds. Preclinical studies support their therapeutic potential, and bioengineering approaches have extended their target range. With their high specificity, rapid action and adaptability, R-pyocins are strong candidates for next-generation precision antimicrobials.
|
|
Scooped by
?
December 10, 11:23 PM
|
As biology grows increasingly data-driven, so too does the field of phage lysins, enzymes that degrade bacterial cell walls and hold promise as alternatives to traditional antibiotics. Five years ago, we introduced PhaLP, a centralized resource for Phage Lytic Protein sequences and associated metadata to support global research efforts. Here, we present PhaLP 2.0, a significantly enhanced database designed to overcome key challenges in the computational study of lysins by integrating the newly identified lysins obtained from thousands of metagenomes. To expand the known diversity of lysins beyond those from cultured phages, we developed SUBLYME, a protein embedding-based machine learning Software designed to Uncover and classify Bacteriophage Lysins in Metagenomic datasets. Using embeddings derived from the prior well-curated protein sequences of the original PhaLP database, we trained support vector machines to distinguish lysins from non-lysins in viromes and classify them as either endolysins or virion-associated lysins. The models achieved an average F1-score of 98% on held-out lysin clusters. SUBLYME enabled the discovery of 743,000 new lysin sequences from EnVhogDB, a virome-derived protein database, increasing the number of known lysin clusters by a factor of 40, from 1,000 to 40,000. PhaLP 2.0 entries were annotated by integrating Pfam functional predictions to the refined delineations obtained with SPAED, an algorithm that leverages the predicted aligned error matrix from AlphaFold predictions to identify domain boundaries. Both SUBLYME and the PhaLP 2.0 database are accessible online at https://github.com/Rousseau-Team/sublyme and http://phalp.ugent.be, respectively. Together, these advances establish PhaLP 2.0 as a comprehensive and scalable portal for the discovery, classification, and sequence analysis of phage lysins, paving the way for future antibacterial applications and evolutionary insights.
|
|
|
Scooped by
?
Today, 3:46 PM
|
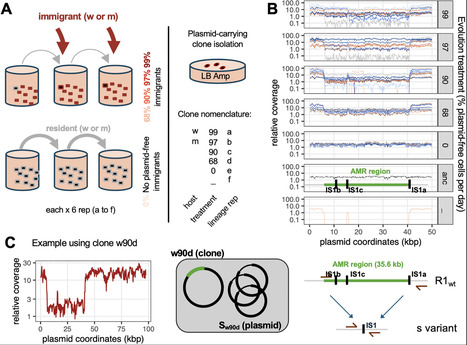
Conjugative plasmids carrying antimicrobial resistance (AMR) genes are critical for the spread of AMR, due to their ability to transmit horizontally between bacterial hosts. We previously observed that during experimental evolution in the presence of abundant susceptible Escherichia coli hosts, the AMR plasmid R1 rapidly evolves variants with increased horizontal transmission due to mutations causing increased plasmid copy number. Yet AMR was progressively lost from the evolving populations. Here, we show that AMR loss was associated with evolution of streamlined plasmids in which the AMR region is spontaneously deleted, making plasmid carriage undetectable by plating on selective antibiotic-containing media. These plasmids transmit both vertically and horizontally more efficiently than the ancestral AMR plasmid, driving AMR extinction in bacterial populations and effectively acting as an intrinsic defence against AMR plasmids. A simple model of plasmid competition further shows that any horizontal or vertical transmission advantage conferred by plasmid streamlining would be enough to drive the displacement of competing AMR plasmids, with a given horizontal transmission advantage leading to faster replacement in conditions favoring horizontal transmission. Our results suggest that within-host plasmid evolution or engineered streamlined plasmids could be exploited to limit the spread of AMR in natural populations of bacteria.
|
|
Scooped by
?
Today, 2:56 PM
|
Recent studies in developing processes using ‘single’ plastic waste for microbial conversion have demonstrated great promise in advancing a circular economy. However, chemical complexity and compositional variability of post-consumer ‘mixed’ plastic waste pose huge challenges to using it as a feedstock for biomanufacturing. Here, we present a process leveraging a synthetic microbial consortium, comprising Rhodococcus jostii strain PET and Acinetobacter baylyi ADP1, enabled by engineering the division of labor. The robust consortium synergistically and stably consumes diverse mixtures of oxygenated compounds, derived from the depolymerization of post-consumer, mixed plastic waste, regardless of the fluctuating plastic waste compositions. We evaluate the upcycling potential of the stable consortium by applying rational metabolic engineering to both specialists, enabling the funneling of these oxygenates into lycopene and lipids. This work highlights the potential of stable microbial consortia to valorize untapped, mixed plastic waste for sustainable biomanufacturing, offering a promising solution to global plastic pollution. The chemical complexity of post-consumer ‘mixed’ plastic waste limits its use as a feedstock for biomanufacturing. Here the authors combine transition-metal-free plastic deconstruction with a microbial consortium platform to upcycle real-world mixed plastic waste into value-added chemicals.
|
|
Scooped by
?
Today, 1:04 PM
|
The application of microbial consortia in biotechnological areas has proven to be much more efficient than that of single microorganisms; however, the main difficulty lies in the large number of communities to be tested. The use of models to predict functional efficiency on a high-throughput scale is key to incorporating greater diversity. The BSocial tool (http://m4m.ugr.es/BSocial.html) assigns a social behavior to each strain based on its contribution to the overall growth of the consortium through a statistical analysis, defining a ‘social consortium’. To determine the effectiveness of the BSocial tool for designing a biofertilizer, the social behavior of 8 plant growth-promoting microorganisms belonging to Azospirillum, Bacillus, Bradyrhizobium, Ensifer and Pseudomonas, as well as 3 plant growth-promoting traits (siderophore production, phosphate solubilisation and indole acetic acid production) of the complete combinatorial (255 communities) were analysed. We selected 3 social consortia (X22, X93 and X149) with a diversity of 2–4 species, two of which presented high performance for more than one plant growth-promoting trait evaluated. Functional stability, following the increase in diversity, was observed in all functions except for siderophore production. Overall, the results show the effectiveness of the BSocial tool in selecting plant growth-promoting consortia to formulate efficient biofertilizers.
|
|
Scooped by
?
Today, 12:19 PM
|
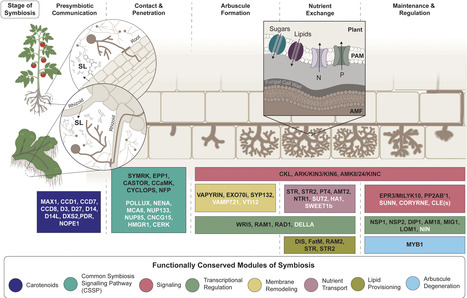
Arbuscular mycorrhizal (AM) symbiosis is an ancient association that played a key role in the adaptation of plants to terrestrial environments. Originating over 400 million years ago at the dawn of land plants, this interaction depends on a core set of conserved genes that enables hosts to establish and maintain symbiotic relationships with AM fungi. The AM symbiotic program includes distinct genetic components for each stage of development, from signal perception to nutrient exchange. Whereas AM host plants have retained key genes dedicated to symbiosis, nonhost lineages have independently lost these genes multiple times over evolutionary history. Recent studies on the liverwort Marchantia paleacea demonstrate that core mechanisms underlying AMF symbiosis are conserved from bryophytes to angiosperms. Comparative genomic studies continue to uncover how symbiosis-specific genes are integrated with broadly conserved cellular machinery to sustain this interaction. Understanding these deeply conserved genetic modules is essential for uncovering the evolutionary foundations of plant–microbe associations and for harnessing their potential in sustainable agriculture.
|
|
Scooped by
?
Today, 11:56 AM
|
Global concern over food waste and plastic pollution highlights the urgent need for sustainable, high-performance materials that can replace petroleum-based plastics. Bacterial cellulose (BC), a biopolymer synthesized through microbial fermentation by Komagataeibacter and related genera, shows exceptional purity, mechanical strength, biodegradability, and structural tunability. Following PRISMA principles, this review analyzed studies from PubMed, Scopus, and Web of Science covering the period 1960–November 2025. Search terms included “bacterial cellulose”, “Komagataeibacter”, “Gluconacetobacter”, “static culture”, “agitated culture”, “in situ modification”, “ex situ modification”, “fermentation”, and “food packaging”. Inclusion and exclusion criteria ensured that only relevant and high-quality publications were considered. The review summarizes major developments in BC biosynthesis, structural organization, and modification approaches that enhance mechanical, barrier, antioxidant, and antimicrobial properties for food packaging. Recent advances in in situ and ex situ functionalization are discussed together with progress achieved through synthetic biology, green chemistry, and material engineering. Evidence shows that BC-based composites can reduce oxygen and moisture permeability, strengthen films, and prolong food shelf life while maintaining biodegradability. Remaining challenges such as high cost, lengthy fermentation, and regulatory uncertainty require coordinated strategies focused on metabolic optimization, circular bioeconomy integration, and standardized safety frameworks to unlock BC’s full industrial potential.
|
|
Scooped by
?
Today, 10:36 AM
|
Methanol is a promising renewable C1 feedstock for biomanufacturing. Native methylotrophs such as Pichia pastoris are considered potential chassis strains for methanol utilization. However, the cytotoxicity of methanol limits its usable concentration, posing a major bottleneck in bioproduction. In this study, we aimed to develop a P. pastoris strain with enhanced methanol tolerance and utilization by adaptive laboratory evolution. Through serial passaging under increasing methanol concentrations, we obtained an evolved strain that exhibited a specific growth rate of 0.101 h− 1 in a 7% (v/v) methanol, a condition under which the wild-type strain failed to grow. Genome resequencing identified the mutations, and the introduction of individual mutations into the wild-type background demonstrated that mutations in PSR1 and BFA1 significantly improved growth under methanol stress. Notably, the fastest-growing isolate, designated PM7a, was capable of growing in a minimal medium containing 5% methanol as the sole carbon source, whereas the wild-type strain was unable to do so. Furthermore, the introduction of the β-carotene biosynthesis pathway into both the wild-type and PM7a strains, cultured in a minimal medium containing methanol as the sole carbon source, resulted in a 4.91-fold higher titer in PM7a. Taken together, our findings demonstrate that ALE facilitates the development of strains with enhanced methanol tolerance, growth, and biosynthetic performance. These findings highlight the potential of the evolved P. pastoris strain as a robust chassis for methanol-based biomanufacturing.
|
|
Scooped by
?
Today, 10:00 AM
|
The fungus Trichoderma reesei is widely employed for industrial enzyme production. However, the mechanisms coordinating enzyme production with amino acid biosynthesis and metabolism remain incompletely understood. Here, we characterized a novel C2H2 zinc finger transcription factor, TrTRC-1, in T. reesei. The deletion of TrTRC-1 enhanced cellulase, xylanase, and extracellular protein production by 9.8–32.4% compared to that in T. reesei Rut C30, while simultaneously stimulating mycelial growth and conidiation. Transcriptomic analysis revealed that the deletion of TrTRC-1 significantly altered the expression of genes associated with primary metabolic pathways, amino acid biosynthesis/metabolism, and carbohydrate-active enzymes. In a 5 L bioreactor, T. reesei ΔTrTRC-1 achieved an FPase activity of 17.35 IU/mL and exhibited robust saccharification efficiency on lignocellulosic biomass. These findings demonstrate that TrTRC-1 serves as a multifunctional regulator in T. reesei, advancing our understanding of the regulatory network that balances enzyme production with cellular metabolism and providing a strategic basis for constructing microbial chassis strains with industrial application prospects.
|
|
Scooped by
?
Today, 9:39 AM
|
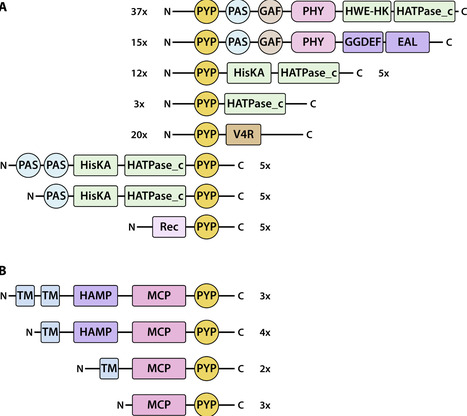
Photoactive Yellow Protein (PYP) is a model system for functional protein dynamics and a prototype of the PAS domain superfamily. It is a bacterial photoreceptor that triggers a range of responses in different bacteria: phototaxis, biosynthesis of photo-protective pigments, and light regulation of biofilm formation. An important gap in knowledge on PYP is the signal transduction chain that guides the initial signal from the photoreceptor to various biological responses. Here, we present an expanded set of 984 PYP homologs, providing information on sequence conservation and variation. We analyze this set of PYPs using two bioinformatics approaches to identify candidate proteins that are functionally related to PYP. First, we identified 153 multi-domain proteins containing PYP and analyzed the domain composition of these proteins. Specific preferences for N- or C-terminal placement of the PYP domain were observed. Second, we identified 113 predicted multi-gene operons containing the pyp gene. These two approaches yielded multiple candidates for proteins in the signal transduction chain associated with PYP, particularly histidine kinase (implying phosphorylation), methyl accepting chemotaxis protein (implying phototaxis), and GGDEF and EAL proteins (implying a role of c-di-GMP and biofilm formation). Some of these candidates were present only in multi-domain proteins and others only in pyp operons. Overexpression of the PYP domain from the MCP-fusion protein from Nitrincola alkalilacustris yielded a protein with an absorbance maximum of 447 nm and an overall photocycle rate of 0.5 s. Our results provide a clear basis for future experimental work on identifying signal transduction partners of PYP.
|
|
Scooped by
?
Today, 9:26 AM
|
Host production of nitric oxide in response to P. aeruginosa results in accumulation of nitrite and nitrate at the infection site, with both utilized for anaerobic respiration to support survival. Nitric oxide and nitrite also act as aerobic respiratory inhibitors. P. aeruginosa must overcome these toxic metabolites alongside self-produced cyanide to persist at the infection site. We previously identified a novel nitrite reductase (NirA) that supports P. aeruginosa virulence in a wide range of infection models. In this work, we demonstrate that mutation of nirA inhibits growth of P. aeruginosa at reduced oxygen tensions in the presence of nitrite or nitrate, with this phenotype shown to be dependent on cyanide. NirA is a siroheme-dependent enzyme, a classical target for inhibition with cyanide. Biochemical characterization confirms that NirA is a novel cyanide-tolerant nitrite reductase, which supports reduction of nitrite in the presence of cyanide. We hypothesise that NirA enables detoxification of nitrite to prevent build-up of multiple respiratory inhibitors and facilitate cyanide-resistant aerobic respiration at low oxygen tensions. Through targeting effectors of these resistance mechanisms, we could promote P. aeruginosa self-poisoning and prevent adaptation to the reduced oxygen environment typically encountered by P. aeruginosa in biofilms and during infection.
|
|
Scooped by
?
Today, 12:14 AM
|
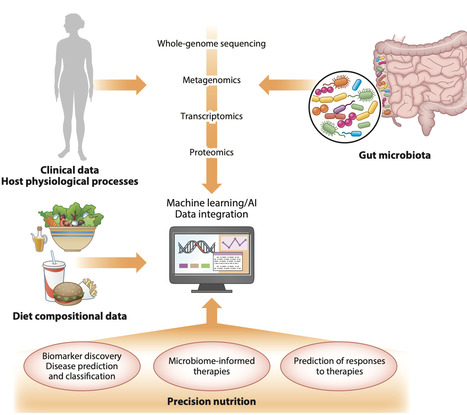
Nutrition plays a fundamental role in shaping human health across the life course, influencing both host physiology and the composition and function of the gut microbiota. In turn, the gut microbiota modulates the effects of dietary intake, creating complex bidirectional interactions with profound implications for metabolic health. Although the concept of personalized nutrition offering tailored dietary advice based on observable traits, environmental factors, and genotype has gained prominence, growing evidence supports the promise of precision nutrition that also considers individual microbiome profiles. This approach is particularly relevant for addressing diet-related conditions such as obesity and type 2 diabetes, where interindividual variability in response to the same diet is well documented. Advances in high-throughput sequencing, metabolomics, and machine learning are driving predictive models that can forecast personalized dietary outcomes. However, methodological heterogeneity, lack of consistency, and limited representation of diverse populations in current studies present significant barriers. Ethical challenges, including data privacy and equitable access to personalized nutrition tools, also warrant urgent attention. To realize the full potential of microbiome-informed nutrition, greater harmonization of research methods, robust validation across large and diverse cohorts, and an interdisciplinary framework are essential.
|
|
Scooped by
?
Today, 12:03 AM
|
Bacterial growth and respiration are fundamental metabolic processes that affect how energy is used and impact carbon sequestration at the ecosystem scale. However, these traits are usually quantified independently, growth is quantified with endpoint biomass measurements while respiration is quantified by monitoring oxygen or carbon dioxide. Because the two physiological traits are collected at different temporal and volumetric scales (hours-to-days for growth versus minutes-to-hours for respiration), reconciling them is challenging and often introduces scale-mismatch bias, obscuring causal links between metabolism and environmental drivers. In this study, we develop a novel method for quantifying the rates of bacterial growth and respiration concurrently from a single dissolved oxygen time series. Our approach introduces a model that couples exponential biomass growth with biomass-specific respiration, enabling the simultaneous inference of both rates in real time. We applied our high throughput method to 15 bacterial strains isolated from natural environments. Our approach yielded growth estimates in close agreement with measurements based on popular methods using optical density or flow cytometry with no evidence of taxon-specific bias. We also tested our approach in quantifying the effects of temperature on respiration, growth and carbon use-efficiency in Pseudomonas sp. Our method yielded typical unimodal thermal response curves for growth and respiration where rates were highest at moderate temperatures, while carbon-use efficiency increased from cooler temperatures, peaked around the thermal optimum, and declined at high temperature. By quantifying respiration and growth simultaneously and in high throughput, our approach effectively enables measurement of microbial metabolic strategies and adaptations to stress. It offers a non-invasive and scalable tool for high throughput phenotyping and studies of environmental perturbations, enabling a new class of trait-based microbial ecology that links cellular physiology to broader ecosystem function.
|
|
Scooped by
?
December 10, 11:27 PM
|
Host specificity of a plant pathogen is defined by its effector complement. However, it remains unclear whether the full complement is required for pathogenicity. Pseudomonas syringae pv. actinidiae (Psa) is an emerging model pathogen of kiwifruit with over 30 functional effectors, providing a unique opportunity to understand how host-mediated selection shapes pathogen evolution. The majority of Psa’s effectors previously appeared nonessential in single knockout contexts. Why, then, does Psa maintain such a large repertoire? We sought to examine effector requirements, redundancies, and repertoire refinement across host genotypes through a mutated effector-leveraging evolution experiment (MELEE), serially passaging competitive populations of effector knockout strains. Competition suggests that all effectors are collectively required for successful virulence, demonstrated by the dominance of wild-type. Host-specific effector requirements identified may further explain the maintenance of this large effector repertoire, providing important insights into the dynamics of effector redundancy following incursions.
|