 Your new post is loading...

|
Scooped by
?
Today, 1:22 PM
|
|
|
Scooped by
?
Today, 1:15 PM
|
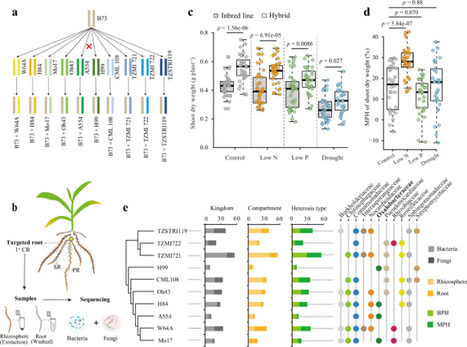
Heterosis, or hybrid vigor, describes the superior performance of F1 hybrids compared to parental inbreds. While soil microbiomes are proposed to influence heterosis, it remains unclear how heterotic plants shape their microbiomes and how interactions relate to stress responses. Here, we investigate the role of rhizosheath formation—the soil tightly adhering to roots—in maize heterosis under nitrogen deprivation. Across sterilization, inoculation, and transplantation experiments, hybrids develop larger rhizosheaths than inbreds, and rhizosheath size associates with biomass heterosis. Rhizosheath-enriched genus Massilia correlates with lateral root density, rhizosheath size, and growth. Untargeted metabolomics and flavone-deficient mutants reveal links between Massilia and flavonoid pathways, while growth promotion by Massilia can also occur independently of host flavones. Metagenomic analysis shows that larger rhizosheaths recruit microbial functions related to nutrient cycling and stress adaptation. These findings identify rhizosheath formation as an integrative trait associated with heterosis and a promising target for breeding resilient crops. This study shows that maize hybrids form larger rhizosheaths than inbreds, which are linked with beneficial microbes such as Massilia. These interactions support root growth, nutrient uptake, and reduced nitrogen loss under stress.
|
|
Scooped by
?
Today, 1:09 PM
|
Ribosomal frameshifting is an important, albeit rare, mRNA decoding mechanism that generally allows the synthesis of a single protein from two different reading frames. +1 frameshifting is commonly presumed to involve re-pairing of the P-site tRNA with the +1 codon. However, in several occurrences in the yeast Saccharomyces cerevisiae, P-site tRNA re-pairing with the +1 codon is impossible. In one model, +1 frameshifting occurs according to a common mechanism involving P-site tRNA movement without re-pairing with the +1 codon. The alternative is a distinct mechanism allowing A-site tRNA acceptance at the +1 codon in the absence of P-site tRNA movement. Here, we experimentally compared all known +1 ribosomal frameshifting sites in S. cerevisiae, including a novel case discovered during this study in LLP1. We identified a conserved RNA secondary structure upstream of the ABP140 frameshifting site that increases frameshifting efficiency. The location of the structure suggests that it creates an mRNA-pulling effect favoring +1 codon in the P-site. Placing the stimulator upstream of various known frameshifting sites revealed that its stimulatory action is selective to those frameshifting sites where P-site tRNA re-pairing is possible, reinforcing the idea of two distinct mechanisms of +1 ribosomal frameshifting.
|
|
Scooped by
?
Today, 1:05 PM
|
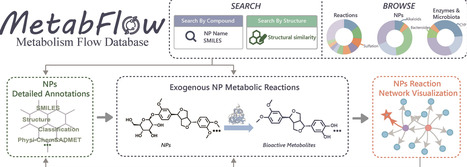
Exogenous natural products (NPs) undergo complex metabolic transformations both in vivo and in vitro, generating structurally diverse metabolites. Due to the involvement of host enzymes and gut microbiota, as well as the structural complexity of NPs themselves, their metabolic processes exhibit unique characteristics. However, existing metabolic transformation information remains fragmented, limiting systematic understanding of their metabolic mechanisms. To address this gap, MetabFlow (https://bddg.hznu.edu.cn/metabflow/), a comprehensive database dedicated to the metabolism of exogenous NPs, was developed. MetabFlow is distinguished by: (a) systematic integration of 7294 curated metabolic reactions covering both in vivo and in vitro transformations, providing a complete knowledge map of exogenous NP metabolism; (b) unique integration of enzyme-catalyzed and microbiota-mediated reactions, highlighting the distinctive metabolic features of NPs; (c) establishment of a structured metabolic network by constructing upstream-downstream reaction relationships, elucidating how exogenous NPs evolve across different metabolic pathways; (d) inclusion of approximately 3200 compounds not indexed in mainstream databases, with detailed structural information, annotations, and complete metabolic profiles. MetabFlow is designed to support research in NP metabolism, drug discovery, and related fields.
|
|
Scooped by
?
Today, 12:53 PM
|
Bacteria constantly adapt to changing environmental conditions through diverse processes that involve numerous regulator and effector proteins. In this regard, small proteins play a significant role in promoting stress adaptation in bacteria. Although they were largely overlooked in early genome annotations, recent technological advances and a growing recognition of their significance have paved the way for the increasing identification and characterization of this intriguing class of proteins. Many small proteins contain a transmembrane domain and are integral to the cytoplasmic membrane. Others interact with and modulate membrane protein complexes. In this review, we focus on the current knowledge of these small membrane proteins, with an emphasis on their interactions, membrane insertion pathways, and toxicity.
|
|
Scooped by
?
Today, 11:29 AM
|
Therapeutically utilized phages should optimally be produced in defined bacterial strains that are free of prophages and virulence factors. However, phage–host interactions in these production strains may be very different from clinical strains. Here, we characterized a lytic Staphylococcus aureus–specific phage vB_SauP_EBHT (EBHT), which had a dramatic change in its host specificity when produced in alternative host 19A2 compared with the original isolation host DSM 104437, even though there were no changes in the phage genome, proteome, structure, or adsorption efficiency. The reason for the altered host range was revealed to be based on different methylation patterns of the EBHT genome by host restriction–modification (R-M) systems in the two hosts. Even though the alternative host 19A2 produced a higher burst size, the host range of the produced phages was narrower. Together, these results illustrate that the most efficient production host may not necessarily be the most optimal one and that bacterial R-M systems should be considered when selecting the optimal phage-production host.
|
|
Scooped by
?
Today, 11:07 AM
|
In recent years, DNA origami technology has advanced rapidly as a groundbreaking method for nano-manufacturing. This technology takes advantage of the unique base-pairing characteristics of DNA, and has significant advantages in constructing spatially ordered and programmable nanostructures. This capability aligns with synthetic biology's core principle of mimicking, extending, and reconstructing natural biological processes by modularly assembling artificial systems. This article provides a comprehensive overview of DNA origami's innovative applications across various domains, including cell membrane surfaces, intercellular communication, intelligent biosensing, and precise gene editing, progressing from the extracellular to the intracellular environment. Finally, this review highlights the synergistic interaction between this technology and cell-free synthetic biology, achieved through the integration of in vitro assembly and cellular regulation, thereby opening new pathways for the rational design of artificial life systems.
|
|
Scooped by
?
November 27, 11:54 PM
|
Understanding how opposing regulatory factors shape gene expression is essential for interpreting complex biological systems. A motivating observation, drawn from cancer epigenetics, is that removing an activating factor can sometimes lead to higher, not lower, expression of a gene that is also subject to repression. This counterintuitive behavior suggests that competition between activators and repressors for limited genomic binding sites may produce unexpected transcriptional outcomes. Prior theoretical work proposed this mechanism, but it has been difficult to test directly in natural systems, where layers of chromatin regulation obscure causal relationships. This paper introduces a fully synthetic, tunable genetic platform in a prokaryotic model system that isolates this competition mechanism in a clean and interpretable setting. The engineered construct contains a target gene with binding sites for both an activator and a repressor, together with a separate decoy region that carries overlapping binding sites for the same regulators. Activator and repressor functions are implemented using CRISPRa and CRISPRi, which permit independent control of regulator expression levels and binding affinities. Using this minimal system, the paper shows that increasing activator expression can reduce expression of the target gene when both regulators are present, consistent with the prediction that additional activator molecules displace the repressor from decoy sites and allow it to more effectively repress the target. By demonstrating how competition alone can invert expected regulatory responses, this synthetic framework provides a validated model for understanding similar paradoxical behaviors in natural regulatory networks and establishes a foundation for future studies in more complex mammalian contexts.
|
|
Scooped by
?
November 27, 11:36 PM
|
Protein-protein interactions are central for understanding biological processes. The ability to predict interaction partners is extremely valuable for avoiding costly, time-consuming experiments. It has been shown that AlphaFold has an unsurpassed ability to accurately evaluate interacting protein pairs. However, a protein can also form homomeric interactions, i.e. interact with itself. We found that AlphaFold yielded a significantly higher false-positive rate for identifying homodimers than for heterodimers. True Positive Rate (TPR) at 1% False Positive Rate (FPR) drops from 63% for heterodimers to 18% for homodimers. When we investigated the high-scoring false positives, i.e., non-homodimers with high AlphaFold scores when predicted as such, we found that their homologs were enriched for homomultimeric proteins. Using a simple logistic regression model that combines AlphaFold scores with structural and homology information, we increased the TPR (at 1% FPR) to 42 +/- 8% (5-fold cross-validation) from 19%. If we excluded the homology information, we achieved a TPR of 28 +/- 7%, which is still better than using AlphaFold metrics. Availability and implementation: All data are available from Zenodo DOI:\10.5281/zenodo.17738668 and all code from https://github.com/SarahND97/alphafold-homodimers
|
|
Scooped by
?
November 27, 11:24 PM
|
Intraspecies interactions shapes microbial community structure and evolution, yet the mechanisms determining competitive outcomes among closely related strains remain unclear. The soil bacterium Bacillus subtilis is a model for microbial social interactions, where quorum-sensing systems regulate cooperation and antagonism. Here, we take a multifaceted approach to dissect the role of quorum-sensing regulation in competitive fitness. Isolate NCIB 3610 carries a signal unresponsive RapP-PhrP module that alters quorum-sensing control and promotes faster growth. Modelling and mutant analysis demonstrate that the small differences in growth rate conferred by RapP-PhrP3610 are sufficient to drive competitive exclusion. The importance of quorum sensing control is further exemplified by experimental evolution of distinct wild isolates, which revealed recurrent mutations in the sensor kinase comP, which phenocopy complete comP or comA deletions and confer a growth-linked competitive advantage. Key quorum sensing mechanisms are abandoned even in structured microbial communities, where it might be expected that communal traits are favored. Furthermore, a phylogenomic survey of 370 B. subtilis genomes identified disruptive comP mutations in ~16% of isolates. However, growth rate alone does not explain all interaction outcomes as even isogenic strains with equivalent doubling times differ in competitiveness. Transcriptomic profiling and validation experiments implicated a type VII secretion system toxin as an additional effector. These findings reveal that disruption of quorum-sensing pathways, whether naturally or through selection, provides a rapid route to competitive advantage, highlighting a fundamental trade-off between communal signalling and individual fitness in microbial populations.
|
|
Scooped by
?
November 27, 11:08 PM
|
Evolution of prokaryote genomes appears to be defined by the interplay of selection for genome streamlining, deletion bias and selection for functional diversification. The previously observed overall positive correlation between the strength of selection, measured as the ratio of non-synonymous to synonymous nucleotide substitutions (dN/dS), points to diversification as the primary factor of prokaryote genome evolution. Here, we investigated the interplay between genome size and selection pressure by analyzing an expanded collection of closely related prokaryotic genomes, evaluating genome-wide selection by measuring dN/dS by using an accurate, phylogeny-based method and decomposing the resulting values into lineage-specific and gene-specific components. These analyses reveal a pronounced heterogeneity in the relationship between genome size and the strength of selection across the diversity of prokaryotes. Most bacteria display a positive correlation consistent with selection for diversification, whereas all analyzed archaeal lineages show strong negative correlation which is the signature of streamlining. These findings indicate that the selection regimes broadly vary across the diversity of prokaryotes rather than following a single, universal pattern. Genome streamlining, selection for functional diversity and drift in small populations are all important factors of evolution, their relative contributions depending on the population genetics and ecology of a given lineage.
|
|
Scooped by
?
November 27, 3:10 PM
|
Cancer treatment mediated by bacteria, also known as Bacteria-mediated cancer therapy (BMCT), has emerged as a promising strategy that overcomes several limitations of conventional cancer treatments by exploiting the natural tumor-targeting ability of bacteria. Among them, Salmonella typhimurium has gained particular attention due to its intrinsic capacity to colonize hypoxic and nutrient-deprived regions of tumors, secrete cytotoxins, and activate host immune responses. This review, along with summarizing these mechanisms, uniquely integrates the diverse anticancer mechanisms of S. typhimurium, such as apoptosis and autophagy induction, immune modulation, nutrient competition, and tumor colonization, which collectively contribute to tumor regression. We discuss the recent advances in metabolic engineering and synthetic biology to provide a unified perspective on how engineered strains achieve enhanced specificity, biosafety, and controlled intratumoral payload delivery. We also critically evaluate the current limitations and translational challenges of BMCT, emphasizing that bacteria-based therapies only complement and not replace existing cancer treatments.
|
|
Scooped by
?
November 27, 2:59 PM
|
The human gut microbiome contains numerous proteins whose functions remain elusive yet are pivotal to host health. Sequence-based methods often falter when attempting to infer functions within this microbial proteome due to evolutionary divergence. To address this challenge, we develop the Human Gut Microbial Protein Structure Database, which incorporates ∼2.7 million predicted protein structures. Our findings reveal that structural analogy enhances the annotation of phage proteins. We detail the structural diversification of phage endolysins and confirm their potential in eliminating gut pathobionts. Furthermore, our structure-guided approach is effective in the identification of microbial-host isozymes. By employing structural alignments, we identify previously unrecognized bacterial enzymes involved in melatonin biosynthesis. Finally, we present an alignment-free method, dense enzyme retrieval, based on structure-encoded protein language models for ultrafast and sensitive detection of remote homologs. Our research underscores the value of computational structural genomics in elucidating the functional landscape of the human gut microbiome.
|
|
|
Scooped by
?
Today, 1:21 PM
|
The outcome of evolution sometimes appears to be predictable, as in the evolution of the same characteristics independently in convergent evolution. Other times, evolution's path depends on starting conditions or chance events, and some forms evolve just once and never appear again. Is convergence, and by implication, predictability, a common characteristic of the evolutionary process? We aimed to answer this question by using an evolutionary video game titled Project Hastur as a study system. In this video game, the enemies evolve traits to help them combat the player's strategy. We determined whether the same environmental pressures (in this case, player strategy) lead to predictable evolution. We conducted a series of experiments with three different playing styles and four different evolutionary treatments with varying kinds of selection pressure, each with several replicates. For each replicate, enemies from the first and final generations were categorized into types using k-means clustering. The Euclidean distance between the cluster centroids and the sum of squares values were recorded for each replicate and compared among evolutionary treatments using Wilcoxon rank sum tests. Evolutionary predictability was evaluated using permutation tests. We found that fitness functions in Project Hastur led to incredible, but unpredictable, diversity. The fitness landscape changes between replicates, even within the same experimental treatment and regardless of player strategy, resulting in enemies with an unpredictable array of trait values.
|
|
Scooped by
?
Today, 1:12 PM
|
RNA-targeted degradation technologies offer significant promise for treating diseases by selectively disrupting gene expression. However, a robust method to specifically, efficiently, and programmability degrade targeted RNAs in mammalian cells is still in demand. Here, we present a versatile platform, dCas13d-directed chaperone-mediated autophagy (dCasCMA), which integrates the precise targeting capabilities of dCas13/CRISPR with the degradation efficiency of chaperone-mediated autophagy (CMA) to achieve efficient degradation of specific RNAs. By combining dCas13d with a CMA-targeting motif and customizable guide RNA (gRNA), the platform allows for accurate targeting of both exogenous and endogenous RNAs in cells. Moreover, the incorporation of multiplexed gRNA expression arrays enables the simultaneous degradation of multiple RNA targets during viral pathogenesis in live cells and in vivo. Our findings emphasize the platform’s modular design, which enables flexible combinations of dCCTM components with user-defined gRNA sequences. This versatility positions it as a promising tool for developing innovative therapies for various diseases. RNA-targeted degradation holds therapeutic potential but still needs more efficient tools. Here, authors develop dCasCMA, a platform that combines CRISPR targeting with a cellular degradation mechanism to efficiently degrade specific RNAs in live cells and in vivo, enabling multi-target therapies.
|
|
Scooped by
?
Today, 1:06 PM
|
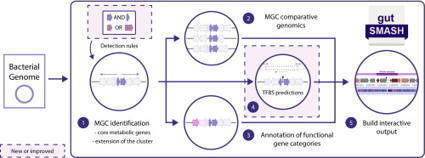
Microbiota-derived metabolites serve as key messengers mediating host–microbe and microbe–microbe interactions, often through specialized primary metabolic pathways. gutSMASH was initially developed to systematically identify the metabolic gene clusters (MGCs) that encode these pathways in anaerobic gut microbial genomes. Here, we present gutSMASH 2.0, a major update that significantly expands its functionality. This version introduces 14 new detection rules covering 12 additional types of MGCs. The comparative genomics framework was enhanced with 26 experimentally validated MGCs and 15,024 gene clusters from the Cultivated Genome Reference 2 (CGR2) collection. Furthermore, gutSMASH 2.0 integrates transcription factor binding site prediction using LogoMotif’s methodology, enabling investigation of MGC regulatory elements. Together, these improvements make gutSMASH a more powerful tool for automated discovery and analysis of niche-determining metabolic pathways in the gut microbiome. gutSMASH 2.0 is freely available at https://gutsmash.bioinformatics.nl/.
|
|
Scooped by
?
Today, 1:01 PM
|
Chemical Entities of Biological Interest (ChEBI) is a high-quality, manually curated, and open-access database and ontology of chemical entities available online at https://www.ebi.ac.uk/chebi/. The chemical entities in question are either naturally occurring compounds or synthetic compounds that play a vital role in the processes of living organisms. ChEBI was launched in 2004, and over the years the original codebase has become increasingly difficult to maintain. Here, we describe the complete overhaul and modernization of ChEBI’s infrastructure, including its codebase and associated tools (website, web services, and submission tool) to ensure the continued availability and growth of this critical resource for the global bioinformatics community and beyond. The infrastructure overhaul also enabled us to introduce new features and capabilities into ChEBI as well as to update or deprecate redundant ones.
|
|
Scooped by
?
Today, 12:48 PM
|
Autonomous bioluminescence systems─genetically encoded platforms that integrate luciferase enzymes with complete substrate biosynthetic pathways─have emerged as transformative tools for real-time, noninvasive imaging in living systems. Unlike conventional substrate-dependent bioluminescence, these systems provide continuous light emission without external substrates, enabling long-term monitoring with minimal phototoxicity compared to the use of fluorescence. Here, we present a critical perspective on recent advances in the two best-characterized autonomous systems: bacterial and fungal bioluminescence systems. We assess their molecular mechanisms, protein engineering strategies, and emerging applications in single-cell imaging, multicolor biosensing, and whole-organism monitoring. By comparing their strengths and limitations, we highlight persistent challenges, such as low quantum yield in bacterial bioluminescence and substrate availability constraints in fungal bioluminescence, and discuss strategies to address them─including AI-guided mutagenesis, de novo protein design, and metabolic pathway optimization. We conclude by outlining application-driven design targets for the next generation of autonomous bioluminescent systems in biomedical research, environmental monitoring, and synthetic biology.
|
|
Scooped by
?
Today, 11:19 AM
|
An enzyme switch, termed “Switchbody”, is developed by fusing an antibody with a fragment of a split enzyme for the precise regulation of enzyme activity in response to an antigen. A luciferase-based Switchbody is engineered by fusing the NanoLuc luciferase fragment HiBiT to the N-terminus of an antibody. The enzyme activity of the Switchbody increases upon the addition of an antigen in a dose-dependent manner in the presence of the complementary fragment LgBiT and its substrate furimazine, demonstrating the potential of the luciferase-based Switchbody as a biosensor. As its working principle, ELISA shows that the interaction between HiBiT and LgBiT is facilitated by antigen binding. Moreover, X-ray crystallography and NMR reveal the heterogeneous trapped state of the HiBiT region and an increasing motility of HiBiT region upon antigen binding, respectively. MD simulations and luminescence measurements show that antigen disrupted the trapping of HiBiT in the antibody, enabling its release. By applying this “Trap and Release” principle to Protein M, an antibody-binding protein, label-free IgG antibodies are successfully converted into bioluminescent Switchbodies. This adaptable Switchbody platform has the potential to expand switching technology beyond luciferase to various other enzymes in the future.
|
|
Scooped by
?
Today, 12:01 AM
|
CRISPR-associated base editors have been established as genome editing tools that enable base conversions in targeted DNA sequences, without generating double-strand breaks. Here, we describe the development of new base editors based on CRISPR-Cas12f1, a miniature Cas protein of only 422 amino acids. Chimeric constructs have been generated by fusing a catalytically inactive dCas12f1, to either a cytosine deaminase or an adenine deaminase. Using these synthetic fusion proteins, systematic analyses have been performed on base editing of a target sequence on a plasmid in Escherichia coli. Interestingly, apart from the previously described base editing of the displaced non-target DNA strand, we also observed efficient editing of the target DNA strand. This effect was not observed for Un1Cas12f1 BEs. In addition to the small size of AsCas12f1 base editors, its unique editing profile makes it a valuable addition to the CRISPR-Cas toolbox.
|
|
Scooped by
?
November 27, 11:43 PM
|
Polyethylene Terephthalate (PET) is an extensively used plastic whose durability and resistance to degradation contribute to growing environmental pollution and concerns. Enzymatic PET degradation, particularly via PETase from Ideonella sakaiensis, has emerged as a sustainable approach due to its ability to depolymerize PET under mild conditions. While research has largely focused on enhancing the enzyme′s thermal stability through distal mutations, less attention has been given to active-site engineering aimed at directly improving catalytic efficiency. Here, we used an automated in silico protein engineering platform called Gene Discovery and Enzyme Engineering (GDEE), designed to systematically explore mutations at the active site. By leveraging FastPETase (FP) as scaffold, we perform a high throughput generation of thousands of variants, evaluated them via docking studies with a PET substrate analogue, and ranked candidates based on binding affinity and catalytic geometry. We identified S238Y as a key mutation that enhanced PET film degrading performance at 40 °C when inserted in two of the most active PETase variants reported to date: 2.2-fold increase in the FP scaffold and 3.4-increase in the ThermoStable-PETase (TSP) background. Compared to wild type PETase, FP S238Y showed a 14.8-fold increase in bulk activity, translating into 9.4-fold more TPA and 20-fold more MHET by UPLC, while TSP S238Y reached a 25.8-fold increase (14.4-fold more TPA and 42.6-fold more MHET). This mutation also enhanced catalytic efficiency and resistance to enzyme concentration inhibition, especially in the TSP scaffold. Molecular dynamics confirm position 238 as a relevant modulator of ligand stabilisation. These findings underscore the potential of targeted active-site engineering, combined with structure-guided prediction, to accelerate the development of efficient mesophilic biocatalysts for plastic waste remediation.
|
|
Scooped by
?
November 27, 11:32 PM
|
Accessibility of the ribosome binding site (RBS) plays an outsized role in bacterial mRNA decay and translation. Antagonistic mRNA sequences that reduce accessibility and regulate expression have been widely documented near the RBS. To determine whether such sequences are also the primary effectors of expression when placed far from the RBS, we measured impacts of all possible 8-nucleotide substitutions (65,536 variants) at different positions in mRNA in Bacillus subtilis. While the vast majority of substitutions negligibly affect RNA levels, pyrimidine-rich substitutions resembling the anti-Shine-Dalgarno (aSD) sequence exhibit strong inhibitory effects. Even several hundred nucleotides downstream of the RBS, these aSD-like sequences base-pair with the RBS, promote RNA decay, and inhibit translation initiation. We find aSD-like sequences to be depleted throughout endogenous genes, likely due to selective pressure for expression. Taken together, our findings reveal widespread long-range RNA intramolecular interactions in vivo and uncover a key constraint on gene sequence evolution.
|
|
Scooped by
?
November 27, 11:19 PM
|
Horizontal gene transfer introduces foreign DNA that can disrupt cellular processes and is therefore subject to xenogenic silencing by nucleoid-associated proteins such as H-NS and Hha. In Enterohaemorrhagic Escherichia coli (EHEC), prophages make up a large fraction of the accessory genome and encode many virulence factors, yet to be expressed they must overcome this silencing. We identify a prophage-encoded small RNA (sRNA), HnrS, that functions as an anti-silencing factor by targeting the H-NS paralogue Hha. HnrS is a short (66-nt) sRNA present in multiple copies (up to nine) in EHEC and Enteropathogenic E. coli (EPEC) genomes and is enriched in E. coli strains that carry the locus of enterocyte effacement (LEE+). We show that HnrS directly base-pairs with the ribosome-binding site of the hha mRNA, repressing its translation and thereby reducing Hha-enhanced H-NS silencing. This counter-silencing de-represses the LEE type III secretion system T3SS and concomitantly represses motility. Transcriptomic profiling further revealed that HnrS indirectly activates genes involved in nitrate/nitrite respiration and nitric oxide resistance, metabolic pathways that contribute to survival in the inflamed gastrointestinal tract. Deletion of hnrS reduced expression of nitrate reductase genes and impaired actin pedestal formation on host epithelial cells. Our results indicate that prophage-encoded, multicopy hnrS provides a counter-silencing mechanism that reduces Hha–H-NS repression at specific virulence loci. This likely enables expression of horizontally acquired genes without broadly disrupting the core H-NS regulon. HnrS illustrates how mobile genetic elements deploy sRNAs to counteract xenogenic silencing and promote virulence gene expression, enhancing colonisation of the host.
|
|
Scooped by
?
November 27, 3:20 PM
|
IscB, as the putative ancestor of Cas9, possesses a compact size, making it suitable for in vivo delivery. OgeuIscB is the first IscB protein known to function in eukaryotic cells but requires a complex TAM (NWRRNA). Here, we characterize a CRISPR-associated IscB system, named DelIscB, which recognizes a flexible TAM (NAC). Through systematically engineering its protein and sgRNA, we obtain enDelIscB with an average 48.9-fold increase in activity. By fusing enDelIscB with T5 exonuclease (T5E), we find that enDelIscB-T5E displays robust efficiency comparable to that of enIscB-T5E in human cells. Moreover, by fusing cytosine or adenosine deaminase with enDelIscB nickase, we establish efficient miniature base editors (ICBE and IABE). Finally, we efficiently generate mouse models by microinjecting mRNA/sgRNA of enDelIscB and enDelIscB-T5E into mouse embryos. Collectively, our work presents a set of enDelIscB-based miniature genome-editing tools with great potential for diverse applications in vivo. Cas9-based genome editing tools face challenges for efficient in vivo delivery due to their large size. Here, the authors present a set of compact genome-editing tools engineered from a CRISPR-associated IscB system which exhibit robust editing efficiency in human cells and mouse embryos.
|
|
Scooped by
?
November 27, 3:09 PM
|
Plants are critical for sustaining human life and planetary health. However, their potential to enable humans to survive and thrive beyond Earth remains unrealized. This Viewpoint presents a collective vision outlining priorities associated with plant science to support a new frontier of human existence. These priorities are drawn from the International Space Life Sciences Working Group (ISLSWG) Plants for Space Exploration and Earth Applications workshop, held at the European Low Gravity Research Association (ELGRA) conference in September 2024. First, we highlight transformative advances gained from using the ‘laboratory of space’ in understanding how plants respond to gravity and other stressors. Second, we introduce a new crop Bioregenerative Life Support System (BLSS) readiness level (BRL) framework – extending the existing Crop Readiness Level (CRL) – to assist in overcoming challenges to establish resilient, sustainable crop production. Materializing the vision of plants as enablers of space exploration will require innovative approaches, including predictive modeling, synthetic biology, robust Earth-based analogue systems, and reliable space-based instruments to monitor biological processes. Success relies upon a unified international community to promote sharing of resources, facilities, expertise, and data to accelerate progress. Ultimately, this work will both advance human space exploration and provide solutions to enhance sustainable plant production on Earth.
|































The Lys-Phe-Glu-Arg-Gln (KFERQ, CTM1) sequence is recognized as a classic targeting motif for CMA pathway23. This sequence can be identified by heat shock chaperone proteins, such as heat shock cognate 70 kDa protein (HSC70), in the cytoplasm. Once recognized, the KFERQ-containing biomolecules are transported to the lysosome via lysosome-associated membrane protein 2 A (LAMP2A), facilitating their degradation within the lysosomal environment.