Your new post is loading...

|
Scooped by
?
Today, 1:04 AM
|
Long-term natural evolution selects glycolysis as the major metabolic mode for rapid cell growth, which however lacks sufficient NADPH supply to dive the biosynthesis of reduced chemicals such as free fatty acids (FFAs). Engineering energy economical pathway for chemical overproduction always compromises cellular fitness due to the rigidity of cellular metabolism. Here, we successfully replaced glycolysis metabolism with an optimized pentose phosphate pathway (PPP) in an industrial yeast Ogataea polymorpha, for the first time, which enabled a higher energy generation efficiency than glycolysis and a balanced supply of ATP and NADPH. More importantly, we discovered a global carbon metabolism regulator CMR that drives metabolic flux toward glycolysis for energy generation, and its disruption relieved the tight regulation of metabolic flux distribution and significantly enhanced the efficiency of cellular energy generation, which significantly boosted the FFA production by 63% in a FFA overproducing chassis. The final engineered yeast produced FFAs at a titer of 41.7 g/L, the highest titer reported by microbial fermentation. Our work provides valuable insights into the metabolic regulation mechanisms and a feasible approach for constructing energy efficient metabolism for chemical overproduction.
|
|
Scooped by
?
Today, 12:59 AM
|
Microbial strains engineered for high-titer ethanol production remain limited in achievable titers compared to native producers such as Zymomonas mobilis. A central unresolved question is why fermentation ceases in such engineered strains before maximum titers are reached. Here, we integrate metabolite profiling with thermodynamic analysis to examine this phenomenon in engineered Escherichia coli and Thermoanaerobacterium saccharolyticum compared with Z. mobilis. In the engineered strains, fermentation cessation coincided with marked pyruvate accumulation. Max-Min Driving Force (MDF) analysis indicated that the pyruvate kinase step in glycolysis approaches thermodynamic equilibrium under physiological metabolite concentrations, reducing the driving force for the pyruvate-to-ethanol branch. Relaxing constraints on pyruvate and related metabolites restored positive MDF values, implicating thermodynamic limitations as the underlying constraint. By contrast, Z. mobilis maintained positive MDF scores without metabolite accumulation, underscoring an organism-specific strategy that avoids this limitation. These findings establish a systems-level framework linking metabolite concentrations to pathway thermodynamics and highlight opportunities for improving microbial performance in ethanol and other bioproduction contexts.
|
|
Scooped by
?
Today, 12:53 AM
|
Coenzyme Q10 biosynthesis in Escherichia coli is constrained by kinetic mismatches between precursor synthesis and methylation, alongside bioenergetic uncoupling. We implemented an optogenetic phase-control strategy integrating dynamic light induction, ribosome binding site (RBS) engineering, and real-time membrane potential (ΔΨ) feedback. Temporal coordination of 1-deoxy-D-xylulose-5-phosphate synthase (DXS) and UbiG methyltransferase (UbiG) via a 6-h phase delay reduced methylglyoxal shunt flux by 41 ± 3% (p < 0.01) through enhanced precursor channeling. Membrane hyperpolarization to − 90 ± 2 mV (relative to − 70 mV in controls) triggered voltage-gated UbiG membrane localization (62 ± 3%) and ATP-driven S-adenosylmethionine regeneration, increasing methylation efficiency 2.3-fold. Multivariate modeling identified ΔΨ and acetate as critical control parameters, enabling optimized fermentation (dissolved oxygen (DO) 15–20%, pH 6.7–6.9). The engineered strain achieved 0.63 ± 0.07 g/L CoQ10 in 5-L bioreactors—a 4.3-fold improvement over the static control strain (0.15 ± 0.02 g/L)—with 82.5% carbon efficiency and 25.8% glycerol-to-product yield. This work establishes bioenergetically coupled temporal control as a scalable paradigm for membrane-bound isoprenoid biomanufacturing.
|
|
Scooped by
?
Today, 12:30 AM
|
Advances in next-generation sequencing technologies have vastly expanded the availability of diverse genomic, epigenomic, and transcriptomic data, presenting the opportunity to develop a general AI model that integrates comprehensive genomic knowledge into a unified model. Unlike previous predictive models, which are typically specialized to certain tasks, our general AI model unifies a wide range of genomic modalities, such as nascent RNA and ultra-high-resolution chromatin organization, within a multi-task architecture. Using ATAC-seq and DNA sequences as inputs, we incorporated diverse genomic modalities as output, and the model exhibits strong generalizability across different cell types and tissues in all tasks we trained. It accurately predicts gene-level transcription measured by various nascent RNA assays, and effectively captures enhancer-associated transcription. Additionally, it accurately captures the potential functions of non-coding genetic variants and regulatory elements. Additionally, we extended the model trained on human data to a mouse general model, achieving accurate predictions of genomic modalities, such as high-resolution chromatin contact maps with limited data availability, which are further validated using an established mouse inner-ear study. This comprehensive approach offers a powerful tool for understanding genome regulation in both human and mouse species.
|
|
Scooped by
?
November 24, 11:44 PM
|
CRISPR activation is a powerful tool to upregulate a vast array of genes in many different contexts. However, there are few dynamic CRISPR transcriptional programs, which limit its usage in the creation of living biosensors, self-regulating microbial factories, or conditional therapeutics. Here, we address this limitation by embedding a molecular switch directly into a guide RNA to create a combined sensor–actuator called a metabolite-responsive scaffold RNA (MR-scRNA). We demonstrate the regulatory potential for MR-scRNAs by conditionally activating genes in three different kingdoms of life. We create MR-scRNAs responsive to two distinct metabolites, theophylline and tryptophan, by swapping the molecular switch used. MR-scRNAs respond quickly in a dose-dependent manner specifically to their target metabolite and enhance biochemical production when used as a dynamic regulator of pathway enzyme expression. The broad functionality and ease of design of the MR-scRNAs offer a promising tool for dynamic cellular regulation.
|
|
Scooped by
?
November 24, 11:37 PM
|
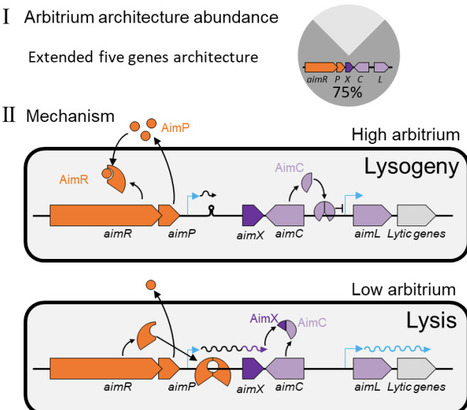
Many temperate Bacillus phages utilize the arbitrium signaling system to control lysis/lysogeny decisions. While the function of the arbitrium signal AimP and its receptor AimR are well known, it is unclear how they control lysis in most arbitrium systems. Here, we show that a large majority of arbitrium systems are embedded in an extended module with three additional components; A small antirepressor protein (AimX), the phage repressor (AimC) and an adjacent cro-like protein (AimL). AimR-dependent activation of AimX is necessary for lysis both during infection and lytic induction. Molecular analysis suggests that AimX directly binds AimC and prevents its oligomerization and binding to its regulated aimL promoter. Our work therefore uncovers the main mechanism by which arbitrium systems regulate lysis and point to the central role of small proteins in phage decision making.
|
|
Scooped by
?
November 24, 11:24 PM
|
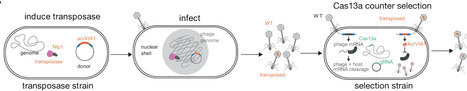
Bacteriophages have genomes that span a wide size range, are densely packed with coding sequences, and frequently encode genes of unknown function. Classical forward genetics has defined essential genes for phage replication in a few model systems but remains laborious and non-scalable. Unbiased functional genomics approaches are therefore needed for phages, particularly for large lytic phages. Here, we develop a phage transposon sequencing (Tn-Seq) platform that uses the mariner transposase to insert an anti-CRISPR selectable marker into phage genomes. CRISPR-Cas13a-based enrichment of transposed phages followed by pooled sequencing identifies both fitness-conferring and dispensable genes. Using the Pseudomonas aeruginosa-infecting nucleus-forming jumbo phage ΦKZ (280,334 bp; 371 predicted genes) as a model, we show that ~110 genes are fitness-conferring via phage TnSeq. These include conserved essential genes involved in phage nucleus formation, protein trafficking, transcription, DNA replication, and virion assembly. We also isolate hundreds of individual phages with insertions in non-essential genes and reveal conditionally essential genes that are specifically required in clinical isolates, at environmental temperature, or in the presence of a defensive nuclease. Phage TnSeq is a facile, scalable technology that can define essential phage genes and generate knockouts in all non-essential genes in a single experiment, enabling conditional genetic screens in phages and providing a broadly applicable resource for phage functional genomics.
|
|
Scooped by
?
November 24, 11:04 PM
|
Advances in CRISPR technologies have led to the use of diverse CRISPR-associated (Cas) proteins in diagnostic applications. Herein, we present a CRISPR/Cas12a2-based amplification-free RNA detection method that exhibits sub-attomolar sensitivity and substrate versatility. Cas12a2, a recently characterized RNA-guided nuclease, uniquely integrates bimolecular recognition through CRISPR RNA (crRNA)-target complementarity and protospacer flanking sequence identification, enabling highly specific trans-cleavage of single-stranded DNA, double-stranded DNA, and RNA. We have optimized key biochemical parameters, including pH, ionic strength, and temperature, to enhance the catalytic efficiency of Cas12a2. Based on the optimal activity conditions of Cas12a2, we have achieved ultra-sensitive viral RNA detection with a limit of detection of 46.7 aM through the strategic design and cooperative activation of crRNAs targeting conserved regions of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome. The diagnostic accuracy of the Cas12a2-based assay has been demonstrated for 26 SARS-CoV-2 variants, and it has further resulted in the definitive diagnosis of 317 clinical samples. This work establishes Cas12a2 as a promising molecular diagnostic tool that provides an amplification-free, rapid, and versatile solution for RNA detection. The adaptability and simplicity of the platform render it particularly well suited for point-of-care applications, paving the way for next-generation CRISPR diagnostics.
|
|
Scooped by
?
November 24, 7:05 PM
|
Kluyveromyces marxianus has high biotechnological potential, particularly because it can utilize a wide range of substrates and grow faster than other yeasts. It has also attracted interest as a prospective probiotic; however, this application is still not well researched. Two K. marxianus strains with promising biotechnological properties and the proven ability to efficiently produce 2-phenylethanol (2-PE) and ethanol were examined in this study to evaluate their potential as probiotics. Yeasts WUT216 and WUT240 exhibited similar survival rates in a simulated human digestive system as the reference K. marxianus strain B0399 (DiarYeast®, LongLife, Milano, Italia). Additionally, the WUT yeasts revealed a high degree of adhesion to cancer and normal cell lines in vitro. Importantly, the WUT strains did not exhibit undesirable characteristics, such as mucolytic and hemolytic activities, and toxic effects on the Caco-2 cell lines in vitro and on Galleria mellonella larvae in vivo. Furthermore, K. marxianus strains displayed attractive probiotic properties, particularly high antioxidant potential, antifungal activity, and significant β-glucan content. This study also showed that the WUT yeasts are sensitive to the commercial antifungals, fluconazole, amphotericin B, and geneticin G418. Overall, the WUT240 and WUT216 exhibit similar probiotic properties to those of B0399. Thus, the WUT yeasts are not only promising producers of 2-PE and ethanol but also attractive probiotic candidates. Additionally, discrepancies in literature reports regarding the analyses conducted were noted, particularly in adhesion and autoaggregation tests.
|
|
Scooped by
?
November 24, 5:19 PM
|
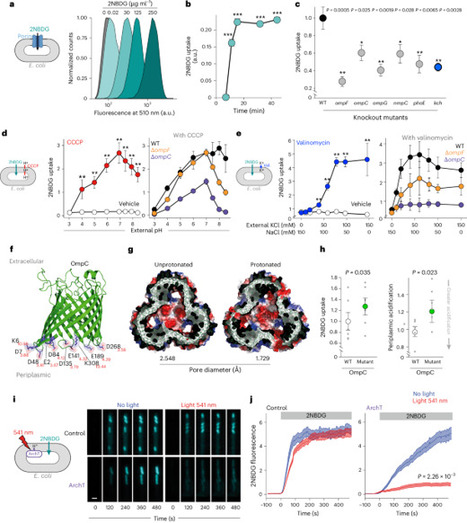
Porins mediate the passage of hydrophilic nutrients and antibiotics across the outer membrane but might contribute to proton leak from the periplasm, suggesting that their conductance could be regulated. Here we show, using single-cell imaging, that porin permeability in Escherichia coli is controlled by changes in periplasmic H+ and K+ concentration. Conductance through porins increases with low periplasmic H+ caused by starvation, promoting nutrient uptake, and decreases with periplasmic acidification during growth in lipid media, limiting proton loss. High metabolic activity during growth in glucose media, however, activates the inner membrane voltage-gated potassium channel, Kch, increasing periplasmic potassium and enhancing porin permeability to dissipate reactive oxygen species. This metabolic control of porin permeability explains the observed increase in ciprofloxacin resistance of bacteria catabolizing lipids and clarifies the impact of mutations in central metabolism genes on drug resistance, identifying Kch as a therapeutic target to improve bacterial killing by antibiotics. The permeability of bacterial porins is dynamically regulated by periplasmic proton and potassium concentrations, altering antibiotic resistance.
|
|
Scooped by
?
November 24, 4:39 PM
|
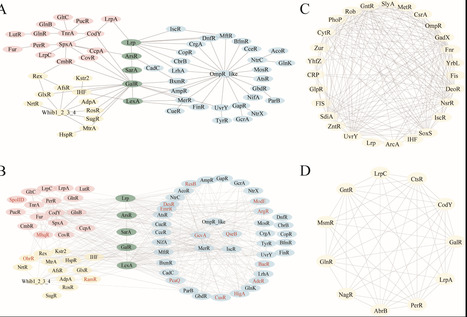
Global regulators (GRs) are key transcription factors that orchestrate the expression of multiple genes, playing essential roles in stress responses, virulence, secondary metabolism, and antibiotic resistance—traits that make them powerful tools for synthetic biology applications. However, conventional approaches often fail to detect remote homologs and novel GR types, limiting our understanding of their regulatory diversity and evolutionary dynamics across prokaryotes. Here, we present a large-scale, protein language model-driven framework to systematically chart the global regulatory landscape across 14,800 bacterial and archaeal type strain genomes—the most taxonomically diverse prokaryotic data set analyzed to date. Using a deep learning-based GR identification model trained on 74,872 curated GR sequences, we systematically identified over 270,000 GR-like proteins, including 173,256 homologs of 214 experimentally validated GR types, 52 putative GR types, and 76,113 proteins of unknown function. This model demonstrated high sensitivity and generalization capability, enabling the discovery of remote homologs and cryptic regulators beyond the reach of similarity- or domain-based methods. This expanded GR catalog revealed lineage-specific distribution patterns, functionally diverse regulons with both conserved and niche-specific targets, and hierarchical cross-regulatory networks with shared and phylum-enriched hubs. By unveiling the hidden diversity and evolutionary structure of prokaryotic global regulators, this landscape of GRs provides foundational insights into microbial gene regulation and offers a powerful toolkit for the rational design of tunable, modular, and orthogonal genetic circuits in synthetic biology.
|
|
Scooped by
?
November 24, 4:24 PM
|
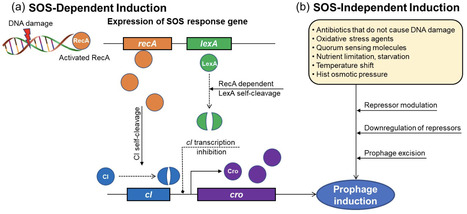
Bacteriophages (phages), the dominant prokaryotic viruses that specifically target bacteria in the human gut microbiome, play a crucial role in maintaining intestinal balance, regulating bacterial populations, and preserving microbial diversity within the gut microbiota. While prophages can enhance bacterial virulence and antibiotic resistance, potentially posing health risks, they also provide beneficial functions, including enhancing host fitness, promoting immune modulation, and contributing to ecosystem resilience, which supports intestinal homeostasis. Human gut microbiota is essential for various physiological functions, including digestion, vitamin synthesis, immune modulation, and protection against pathogens. Dysbiosis, or microbial imbalance, is associated with various disorders such as inflammatory bowel disease, obesity, diabetes, and mental health disorders. Consequently, prophages are important considerations for developing therapies to prevent intestinal diseases. Recently, there has been significant interest in prophage induction in the gut due to its functional impacts on microbial dynamics, gut health, and disease modulation. Prophage induction can be regulated by diet, antibiotics, metabolites, gut health, lifestyle, and intestinal environments. However, compared with lytic phages, prophages remain underexplored, leaving gaps in our understanding of their functions within the gut. Therefore, further research is needed to fully elucidate the complex interactions between phages, prophages, and the gut microbiota, and their effects on health and disease. This knowledge could inform the development of phage-based therapies and improve therapeutic strategies for gut health.
|
|
Scooped by
?
November 24, 4:05 PM
|
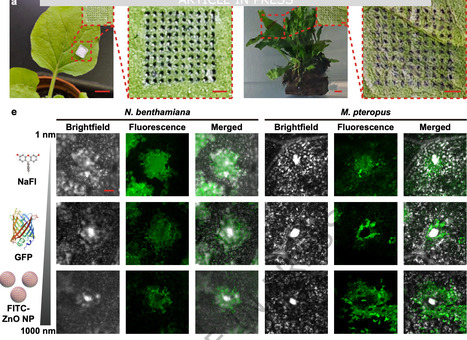
Efficient cargo delivery is essential for plant trait engineering, yet existing methods are often species-specific and ineffective across diverse habitats. Here, we develop core-shell microneedles for targeted delivery of biomolecular cargoes and active microorganisms into both terrestrial and aquatic plants. The microneedle architecture is rationally engineered to resist water exposure and release cargo upon contact with plant interstitial fluid, enabling controlled delivery into tissues and cells. We demonstrate that these core-shell microneedles can efficiently transport diverse cargoes, from nanoscale biomolecules such as functional nucleic acids, proteins, and plant hormones to microscale bioactive Agrobacterium, leading to strong protein expression and enhanced plant growth. Underwater delivery of salt-tolerance genes into submerged freshwater plants further demonstrates the platform’s utility for engineering stress resilience in challenging environments. By facilitating the cellular uptake of diverse cargoes into intact plants across different habitats, this amphibious microneedle strategy offers a versatile cargo delivery tool to advance plant biotechnology and environmental applications. An efficient cargo delivery tool is essential for plant trait engineering. Here, the authors repot a core-shell microneedles for targeted delivery of nucleic acids, proteins, phytohormones, and Agrobacterium to both terrestrial and aquatic plants.
|
|
|
Scooped by
?
Today, 1:02 AM
|
Bioluminescent luciferases have emerged as powerful tools for bioimaging, enabling to image biological systems without external excitation light, reducing thus phototoxicity and eliminating background autofluorescence. Advanced imaging requires luciferases that deliver high photon output for enhanced sensitivity, tunable emission colors for multicolor imaging, and red-shifted emission for effective deep tissue imaging. Here, we introduce LumiFAST, a small tunable luciferase engineered by fusing the bright blue-light emitting NanoLuc with the tunable chemogenetic fluorescent reporter pFAST. pFAST binds and stabilizes the fluorescent state of a variety of synthetic fluorogenic chromophores (also called fluorogens). Its proximity with NanoLuc leads to efficient bioluminescence resonance energy transfer (BRET), enabling customizable red-shifted emission. Thanks to the small size of pFAST, LumiFAST maintains a compact structure, while its modular design allows emission color to be tuned from cyan to green, yellow, orange and red simply by changing the fluorogen. Systematic optimization of the fusion topology and linker length yielded an optimal variant with apparent BRET efficiencies reaching up to 90 %. The red-shifted emission of LumiFAST enables dual-color microscopy imaging when used alongside NanoLuc and allows imaging through thick scattering media. Beyond imaging, our insights into the structural factors governing efficient BRET allowed us to engineer biosensors based on NanoLuc and pFAST for the visualization of protease activity and protein-protein interactions in live cells.
|
|
Scooped by
?
Today, 12:56 AM
|
We introduce BoltzGen, an all-atom generative model for designing proteins and peptides across all modalities to bind a wide range of biomolecular targets. BoltzGen builds strong structural reasoning capabilities about target-binder interactions into its generative design process. This is achieved by unifying design and structure prediction, resulting in a single model that also reaches state-of-the-art folding performance. BoltzGen's generation process can be controlled with a flexible design specification language over covalent bonds, structure constraints, binding sites, and more. We experimentally validate these capabilities in a total of eight diverse wetlab design campaigns with functional and affinity readouts across 26 targets. The experiments span binder modalities from nanobodies to disulfide-bonded peptides and include targets ranging from disordered proteins to small molecules. For instance, we test 15 nanobody and protein binder designs against each of nine novel targets with low similarity to any protein with a known bound structure. For both binder modalities, this yields nanomolar binders for 66\% of targets. We release model weights, data, and both inference and training code at: https://github.com/HannesStark/boltzgen.
|
|
Scooped by
?
Today, 12:46 AM
|
Single-molecule tracking (SMT) is a powerful tool for real-time studies of protein interactions in living cells. Dye-labelled SNAP-tag and HaloTag self-labelling proteins have simplified SMT significantly, due to their superior photophysical properties compared to fluorescent proteins. However, due to their size, fusion of these tags to a protein of interest often results in loss of protein function. We introduce FLORENCE – a universal labelling method for SMT, based on genetic code expansion (GCE). We overcome significant caveats related to recoded strains, vectors, and dyes, and report successful tracking of site-specifically intracellularly labelled proteins in genomically re-coded E. coli. Our findings establish a robust in vivo protein-labelling strategy, expanding the capabilities of SMT as a method to study the dynamics of proteins in living cells. Moreover, we observe that the strain-promoted azide-alkyne click-chemistry reaction occurs as fast as 30 min in live E. coli cells, and can be used as a robust labelling reaction.
|
|
Scooped by
?
November 24, 11:56 PM
|
Core promoters comprise multiple elements whose interaction with RNA polymerase initiates transcription. Despite decades of research, substantial sequence and length variation of promoter elements has hindered efforts to elucidate their function and the evolutionary diversity of transcriptional regulation. Combining massively parallel assays, biophysical modeling, and functional validation, we systematically dissected the promoter architecture upstream of experimentally determined transcription start sites in 49 phylogenetically diverse bacterial genomes (GC content: 27.8%–72.1%). We identified a conserved 3-bp promoter element, termed “start,” which dictates transcription start site selection and enhances transcription. We uncovered a four-region organization within the variable spacer element, whose sequence composition modulates transcription by up to 600-fold. We showed that the discriminator element is conserved in Terrabacteria but diverse in Gracilicutes, the two major bacterial clades. High discriminator sequence diversity in Gracilicutes likely reflects diversifying evolution, enabling promoter-encoded regulation to orchestrate global gene expression in response to growth rate changes. Together, our findings reveal broad conservation of bacterial promoter organization while highlighting regulatory divergence of promoter elements and RNA polymerase between Terrabacteria and Gracilicutes. Sequence and functional similarities between bacterial promoter elements and their archaeal and eukaryotic counterparts further suggest a shared evolutionary origin of promoter architecture.
|
|
Scooped by
?
November 24, 11:41 PM
|
Bacterial flagella are known for facilitating motility to support nutrient acquisition and predator evasion, but can also serve as receptors for phages. Here, we characterize a dual role of flagella during infection of Yersinia enterocolitica by phage X1: functioning as a conduit for phage DNA entry and as a trigger for bacterial defense. X1 employs a “contraction-driven” mechanism, using its contractile tail to inject DNA into Yersinia flagella. Unlike other characterized flagellotropic phages that rely exclusively on flagella or less efficiently on secondary surface receptors, X1 can infect cells via lipopolysaccharides with even higher efficiency. Furthermore, certain Yersinia strains can detect X1 invasion and activate a flagellum-dependent toxin–antitoxin (Flag-TA) system, which requires the flagellar motor stator proteins MotAB to activate the abortive infection response. Our findings reveal a novel phage infection strategy and uncover a new bacterial defense that are both based on the flagellar machinery.
|
|
Scooped by
?
November 24, 11:28 PM
|
Communities of bacteria undergo population bottlenecks which are crucial to their population, ecological, and evolutionary dynamics. However, conventional amplicon sequencing cannot distinguish such demographic shifts between very closely related lineages. Here we describe BarTn7, an optimized method for bacterial lineage tracking through chromosomal integration of phenotypically neutral DNA barcodes with transposon Tn7. By combining conventional conjugative plasmids into a single vector and leveraging a parts-based strategy to optimize delivery to different recipients, BarTn7 increases barcoding efficiency and enables the systematic application of this tool to diverse bacteria. We tested BarTn7 in multiple bacterial species and confirmed a lack of lineage-specific growth effects. We then used BarTn7 to measure the colonization of Panicum virgatum roots under differing phosphate concentrations. Lineage tracking enabled discrimination between unique colonization events and the proliferation of existing bacteria during root colonization, the ratio of which differed between three plant-associated bacterial species. The strategy further allowed the measurement of phosphate-dependent ingress rates of the root endosphere by Paraburkholderia phytofirmans PsJN. We then demonstrated the effectiveness of BarTn7 to detect adaptive mutation(s) and facilitate identification of mutant lineages. Additionally, we demonstrated that BarTn7 is more accurate than conventional 16S rRNA gene sequencing at measuring community composition of a 5-member synthetic bacterial community (SynCom) and compares favorably to shotgun metagenomics. Our results illustrate the utility of BarTn7 as a simple, cost-efficient, and broadly applicable method of measuring bacterial population dynamics at sub-species resolution.
|
|
Scooped by
?
November 24, 11:19 PM
|
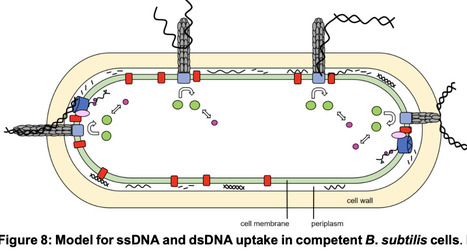
Natural competence describes the ability of several bacterial species to take up DNA from the environment for integration into the host chromosome, allowing the acquisition of novel genetic information. Uptake in many species operates via a dynamic pilus structure that pulls DNA through the cell wall into the periplasm by pilus retraction, followed by transport of one of the two DNA strands into the cytosol by an active membrane transporter. How double stranded DNA with a bending contour of hundreds of base pairs can fit through a narrow pore accommodating a pilus has been a puzzling question. We show that purified major and minor pilins from two Bacillus species, B. subtilis and Geobacillus thermodenitrificans, show efficient binding to double as well as to single stranded DNA with similar affinities. Consistent with uptake, ssDNA accumulates and moves within the periplasm before transport through the cell membrane, with similar dynamics as dsDNA. Thus, Bacillus competence pili are dsDNA as well as ssDNA uptake machines. The binding of pili to ssDNA regions within environmental DNA likely increases the efficiency of competent bacteria to pull extended DNA strands through the cell wall. Uptake of environmental ssDNA may also provide a beneficial source of nutrition for bacteria during stationary.
|
|
Scooped by
?
November 24, 7:27 PM
|
ArsR-based whole-cell biosensors offer sensitive colorimetric detection of arsenite [As(III)], yet their broad reactivity toward Group 15 metalloids—especially antimonite [Sb(III)]—limits field specificity. The recently identified ant operon from Comamonas testosteroni JL40 confers Sb(III)-selective resistance via the efflux ATPase AntA, the metallochaperone AntC, and the regulator AntR, providing genetic parts to suppress Sb(III) cross-talk. We systematically introduced antA, antC, and three antR homologs into an ArsR regulator coupled with a deoxyviolacein reporter chassis (pJ23119-K12). Co-expression of AntA and AntC under a moderate constitutive promoter (PceuR) shifted the Sb(III) limit of detection (LOD) from 0.073 µM to 0.586 µM, with a modest increase in the As(III) LOD to 0.018 µM. Subsequent integration of AntR1 not only maintained the As(III) LOD at 0.018 µM but also unexpectedly amplified the As(III) signal, extending the linear range to 36 nM–37.5 µM (R² = 0.991). It suggests that AntR1 may modulate the transcriptional circuitry via cross-regulation, warranting further mechanistic inquiry. The modified biosensor TOP10/pJ23119-antACR1 exhibited high selectivity for As(III) over divalent metals (Cd, Pb, Cu, Hg, Mn, Mg) and tolerated Sb(III) up to 1 µM. Performance was retained in 90% freshwater and 50% seawater matrices, enabling accurate quantification of 0–2.5 µM As(III) in deionized, tap, surface, and marine samples. By coupling Sb(III)-specific ant efflux/sequestration components with an ArsR-based sensing module, we developed a portable, low-cost biosensor that overcomes longstanding As(III)/Sb(III) cross-reactivity and performs robustly in complex environmental waters.
|
|
Scooped by
?
November 24, 6:43 PM
|
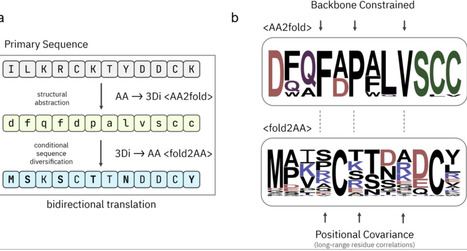
The accuracy of protein structure prediction models such as AlphaFold2 is tightly coupled to the depth and quality of multiple sequence alignments (MSAs), posing a persistent challenge for proteins with few or no identifiable homologs. We present GhostFold, a method for conjuring structure-constrained synthetic MSAs from a single amino acid sequence, bypassing the need for traditional homology searches. Leveraging the ProstT5 protein language model and the 3Di structural alphabet, GhostFold projects a query sequence into a tokenized structural representation and iteratively back-translates to generate an ensemble of diverse, fold-consistent sequences. These synthetic alignments (pseudoMSAs) encode emergent coevolutionary constraints that are sufficient for high-accuracy structure prediction of difficult targets such as orphan proteins and hypervariable antibody loops. GhostFold consistently matches or exceeds the performance of MSA-based and language model-based structure predictors while being computationally lightweight and independent of large sequence databases. Notably, we observe a decoupling of confidence metrics (e.g., pLDDT) from prediction accuracy when using pseudoMSAs, suggesting that AlphaFold2’s internal confidence calibration is strongly influenced by the statistical properties of natural sequence alignments. These results establish that structure-guided synthetic MSAs can functionally substitute for evolutionary data, offering a scalable and generalizable solution to one of the central limitations in computational structural biology. GhostFold represents a shift from passive data mining to intelligent sequence synthesis, redefining how structural priors are encoded in deep learning-based protein folding.
|
|
Scooped by
?
November 24, 4:49 PM
|
The diverse regulatory functions, protein production capacity, and stability of natural and synthetic RNAs are closely tied to their ability to fold into intricate structures. Determining RNA structure is thus fundamental to RNA biology and bioengineering. Among existing approaches to structure determination, computational secondary structure prediction offers a rapid and low-cost strategy and is thus widely used, especially when seeking to identify functional RNA elements in large transcriptomes or screen massive libraries of novel designs. While traditional approaches rely on detailed measurements of folding energetics and/or probabilistic modeling of structural data, recent years have witnessed a surge in deep learning methods, inspired by their tremendous success in protein structure prediction. However, the limited diversity and volume of known RNA structures can impede their ability to accurately predict structures markedly different from the ones they have seen. This is known as the generalization gap and currently poses a major barrier to progress in the field. In this Perspective article, we gauge method generalizability using a new benchmark dataset of structured RNAs we curated from the Protein Data Bank. We also discuss the emergence of deep learning methods for predicting structure probing data and use a new dataset to underscore generalization challenges unique to this domain along with directions for future improvement. Expanding beyond improving predictive accuracy, we review how advances in deep learning have recently enabled scalable and accessible optimization of traditional structure prediction methods and their seamless integration with modern neural networks.
|
|
Scooped by
?
November 24, 4:33 PM
|
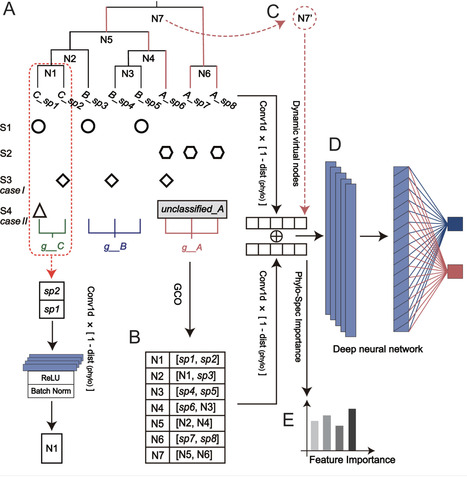
The human microbiome is crucial for health regulation and disease progression, presenting a valuable opportunity for health state classification. Traditional microbiome-based classification relies on pre-trained machine learning (ML) or deep learning (DL) models, which typically focus on microbial distribution patterns, neglecting the underlying relationships between microbes. As a result, model performance can be significantly affected by data sparsity, misclassified features, or incomplete microbial profiles. To overcome these challenges, we introduce Phylo-Spec, a phylogeny-driven deep learning algorithm that integrates multi-aspect microbial information for improved status recognition. Phylo-Spec fuses convolutional features of microbes within a phylogenetic hierarchy via a bottom-up iteration and significantly alleviates the challenges due to sparse data and inaccurate profiling. Additionally, the model dynamically assigns unclassified species to virtual nodes on the phylogenetic tree based on higher-level taxonomy, minimizing interferences from unclassified species. Phylo-Spec also captures the feature importance via an information gain-based mechanism through the phylogenetic structure propagation, enhancing the interpretability of classification decisions. Phylo-Spec demonstrated superior efficacy in microbiome status classification across two in silico synthetic data sets that simulate the aforementioned cases, outperforming existing ML and DL methods. Validation with real-world metagenomic and amplicon data further confirmed the model’s performance in multiple status classification, establishing a powerful framework for microbiome-based health state identification and microbe-disease association. The source code is available at https://github.com/qdu-bioinfo/Phylo-Spec.
|
|
Scooped by
?
November 24, 4:12 PM
|
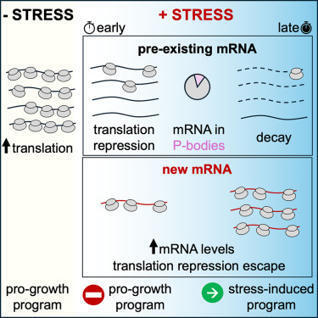
When cells encounter stress, they rapidly mount an adaptive response by switching from pro-growth to stress-responsive gene expression programs. How cells selectively silence pre-existing, pro-growth transcripts yet efficiently translate transcriptionally induced stress mRNA and whether these transcriptional and post-transcriptional responses are coordinated are poorly understood. Here, we show that following acute glucose withdrawal in S. cerevisiae, pre-existing mRNAs are not first degraded to halt protein synthesis, nor are they sequestered away in P-bodies. Rather, their translation is quickly repressed through a sequence-independent mechanism that differentiates between mRNAs produced before and after stress, followed by their decay. Transcriptional induction of endogenous transcripts and reporter mRNAs during stress is sufficient to escape translational repression, while induction prior to stress leads to repression. Our results reveal a timing-controlled coordination of the transcriptional and translational responses in the nucleus and cytoplasm, ensuring a rapid and wide-scale reprogramming of gene expression following environmental stress.
|