Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 6:10 PM
|
Accurate gene prediction remains a major bottleneck in fungal genomics, where lineage diversity and alternative splicing challenge existing ab initio methods. Here, we present geneML, a deep learning-based gene prediction tool tailored to fungal genomes. Across nine reference genomes spanning diverse fungal taxa, geneML improved gene-level F1 score from 64.9 to 67.1 compared to BRAKER3 with protein-based hints, driven by substantially higher recall (69.0 vs. 64.1) at equivalent precision. geneML also remains fast, averaging around 6 minutes per genome on a standard 8-core CPU. A key feature of geneML is its ability to predict alternative transcripts. Compared to Fusarium graminearum Iso-Seq control data, it achieves 41.1% transcript recall and 71.1% precision, outperforming AUGUSTUS (33.8% recall, 48.9% precision), one of the few tools that support isoform prediction. The predicted transcript diversity is consistent with experimentally observed fungal alternative splicing patterns. Reannotation of the curated training dataset further suggests improved biological completeness, with geneML recovering 15.3% more genes containing complete PFAM domains than the reference annotation. These results demonstrate that geneML enables faster, more sensitive, and more biologically informative fungal genome annotation. geneML is available as an open-source command-line tool at https://github.com/hexagonbio/geneML.
|
|
Scooped by
mhryu@live.com
Today, 5:59 PM
|
Rapid quality assessment and multiple assembly comparison are essential steps while assembling new genomes or re-sequencing known ones. Many available tools used for assembly evaluation produce global metrics, representing assembly quality or overall features, most of them working as command line tools that typically act on large data files and produce long detailed result files, where it is not always easy to identify regions of similarity or difference among different chromosome assemblies. ChromoMapperWeb is a new web tool that takes as input nucmer or QUAST output files, quickly identifies similarities and differences between the compared assemblies, and displays them using both table visualizations and pre-arranged or custom graphics. Graphical displays are interactive and allow progressive zoom levels which, in a few steps, move from full genome to very enlarged views, where even small alignment blocks are easily identified. The program, freely accessible through the web server https://chromomapperweb.ceinge.unina.it/, provides an easy-to-use graphical interface, used for experiment planning and interactive evaluation of the results, which include tables and graphical representations of whole genomes, chromosomes, or single blocks.
|
|
Scooped by
mhryu@live.com
Today, 5:53 PM
|
Microbe Decoder is a web server that predicts functional traits of microbes in microbiome sequencing datasets. Sequencing has revealed thousands of organisms in most ecosystems, but the functional traits of many organisms remain unclear. Existing tools can predict names of organisms or their genes, but they rarely predict concrete biological functions (e.g. fermentation or anaerobic growth). Microbe Decoder fills this gap using three complementary tools relying on taxonomy, metabolic networks, or machine learning. These tools accept either names or gene functions as inputs and are integrated into an easy-to-use web app. When tested against data for microbial isolates, Microbe Decoder showed good predictive performance (e.g. balanced accuracy of 0.85). When applied to datasets from the gut, sediment, and sea, it predicted shifts in functional traits over space and time. Microbe Decoder is designed for use with prokaryotes, with the goal of including eukaryotes in the future. By revealing functional traits of microbes in biological systems, Microbe Decoder will advance biology, medicine, and environmental science. Microbe Decoder is available at https://www.microbe-decoder.org/.
|
|
Scooped by
mhryu@live.com
Today, 5:43 PM
|
Fungi are a key component of the microbial community in soils, forming species-rich assemblages in soils of natural ecosystems and managed soils. Their unique physiology, absorptive nutritional mode and growth form as spore-producing filamentous eukaryotes enables them to make specific contributions to a wide range of ecological roles as potent decomposers, versatile mutualists and also destructive pathogens. Fungi have important roles in biogeochemical cycles, for example, in nutrient mineralization, plant nutrient uptake and carbon storage. Positioned at the basis of the soil food web, they are one of the pillars of the flow of carbon and energy through the soil food web. As bottom-up controlled organisms in the soil, largely controlled by their resources, they react sensitively to a range of anthropogenic factors, including climate change and other factors of global environmental change, such as chemical pollution. By influencing the food system and via their role as pathogens and in antifungal resistance, fungi are key players in One Health and planetary health. In this Review, Rillig explores the diversity of fungi in the soil ecosystem, their ecological interactions and diverse ecological roles in terrestrial ecosystems as well as anthropogenic factors that affect soil fungi.
|
|
Scooped by
mhryu@live.com
Today, 5:27 PM
|
The limited availability of structurally specific hydroxylated tryptophan derivatives, whether they are obtained through natural extraction or chemical synthesis, presents challenges for industrial applications. In this study, we report a modular biosynthetic platform in E. coli for producing melatonin and related indoleamines by functionally integrating an animal hydroxylase, an insect decarboxylase, and an avian N-acetyltransferase as well as a plant O-methyltransferase. Following the rational design of key enzymes as well as metabolic pathway optimization, the recombinant strains produced 22.8 g/L of 5-hydroxytryptophan (5-HTP) in a 70 L bioreactor and 6.2 g/L of melatonin in a 5 L bioreactor with exogenous tryptophan feeding. This engineered microbial system establishes an efficient route for manufacturing pharmaceutically relevant tryptophan derivatives and provides a versatile enzymatic toolkit for exploring hydroxylated indole compound biochemistry.
|
|
Scooped by
mhryu@live.com
Today, 4:46 PM
|
Biomolecular condensates have emerged as versatile regulators of plant cellular processes, offering a dynamic and reversible mechanism to coordinate development, stress response, and spatial organization. Through phase separation, these condensates spatially and temporally modulate biochemical reactions, sequester or activate specific proteins and RNAs, and reshape cellular architecture. This review presents a comprehensive and multidimensional framework for understanding biomolecular condensates in plant biology, from their biophysical properties and ensemble dynamics to their roles across diverse cellular compartments, including plasma membranes, cytoskeleton, intracellular compartments, and chromatin. We highlight their functions in growth, environmental sensing, and defense and discuss current challenges in studying their composition, material properties, and context-dependent behaviors. Understanding plant condensates not only deepens our knowledge of plant cell organization and adaptability but also opens new avenues for biotechnological innovation in agriculture.
|
|
Scooped by
mhryu@live.com
Today, 4:38 PM
|
Xenonucleic acids (XNAs) are defined as sugar-modified nucleic acids, which can be broadly categorized into two groups: those that substitute the ribose or deoxyribose for another sugar or sugar derivative, and those that replace the sugar moiety with a non-sugar unit. These chemical modifications confer enhanced stability, binding affinity, and functional versatility beyond the capabilities of DNA and RNA. This comprehensive review examines the fundamental properties, synthesis methodologies, structural characterization, and biomedical applications of XNAs. Major XNA variants, including locked nucleic acids (LNAs), peptide nucleic acids (PNAs), hexitol nucleic acids (HNAs), fluoro-arabino nucleic acids (FANAs), and morpholino oligomers (MOs), exhibit remarkable nuclease resistance and thermal stability. Synthesis approaches range from traditional phosphoramidite chemistry to enzymatic methods utilizing engineered polymerases and innovative hybrid strategies. Sophisticated characterization techniques, including thermal melting analysis, circular dichroism (CD) spectroscopy, nuclear magnetic resonance (NMR), mass spectrometry (MS), and adapted sequencing methods, enable detailed structural and functional analysis. XNAs have achieved significant clinical impact through FDA-approved antisense therapeutics and revolutionized molecular diagnostics via ultrasensitive liquid biopsy technologies. Current challenges include scalable synthesis, effective delivery systems, and comprehensive structure-function understanding. Future perspectives encompass AI-guided design, synthetic biology applications, and expanded therapeutic pipelines, positioning XNAs as transformative tools in precision medicine and biotechnology.
|
|
Scooped by
mhryu@live.com
Today, 12:52 PM
|
RNA interference (RNAi) has emerged as a promising approach to sustainable crop protection. Extensive proof-of-concept studies have led to the approval of the first sprayable plant protection products in the United States and China, with Europe currently evaluating them. Although gene silencing mechanisms are among the most extensively studied processes in molecular biology, with two Nobel Prizes recognizing their discovery, the optimization of delivery systems and field performance remains an area currently undergoing extensive development. The uptake, stability, and efficacy of double-stranded RNA (dsRNA) are influenced by species-specific and environmental factors, introducing variability that must be understood in order to select robust targets, design effective dsRNA, and assess risk. Although these knowledge gaps remain, they are increasingly addressed through systematic experimental and technological advances. This review summarizes the current knowledge on RNAi mechanisms in plants, fungi, and insects, emphasizing the differences in dsRNA uptake and processing between species. We highlight advances in formulations and delivery technologies, discuss how regulatory and ecological questions are being systematically investigated, and present examples of approved products that demonstrate the approach's feasibility and safety. Finally, we outline how remaining uncertainties can be addressed through targeted research and risk-mitigation strategies and how RNAi technologies can be incorporated into comprehensive pest and disease management systems.
|
|
Scooped by
mhryu@live.com
Today, 12:32 PM
|
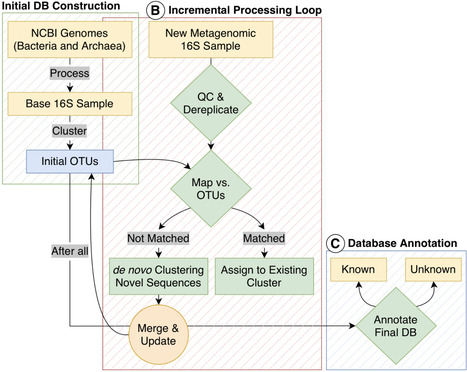
The exponential growth of public metagenomic datasets offers an unprecedented opportunity to explore microbial diversity. However, analyzing this vast amount of data presents significant computational challenges. While shotgun metagenomics provides deep functional and taxonomic resolution, its high cost still limits its application. On the other hand, 16S rRNA gene sequencing remains a cost-effective and widely used alternative, but tools are needed to maximize its discovery potential. Traditional clustering is not scalable, obstructing the creation of a comprehensive and continuously updated catalog of microbial life from 16S data. We developed a reproducible and scalable Snakemake pipeline for the incremental clustering of 16S rRNA amplicons. The workflow begins by constructing a reference database from bacterial and archaeal genomes. It then processes 16S rRNA samples sequentially. For each new sample, sequences are first mapped against the existing cluster centroids. Sequences that match known centroids are assigned accordingly, while unmapped sequences are clustered independently to form novel operational taxonomic units (OTUs). These new centroids are then merged with the existing database, allowing it to grow dynamically without the need for computationally prohibitive all-at-once re-clustering. Our pipeline enables the efficient and continuous expansion of a 16S rRNA cluster database. By processing a large corpus of public 16S rRNA samples, we generated a comprehensive atlas of tens of thousands of OTUs. A significant fraction of these clusters, particularly at the genus and family levels, were classified as unknown. This work provides a powerful, open-source tool for large-scale analysis of 16S rRNA samples. The incremental clustering strategy overcomes the scalability limitations of traditional methods, allowing researchers to leverage public data and discover novel microbes in their own microbiome samples.
|
|
Scooped by
mhryu@live.com
Today, 12:19 PM
|
Sponge RNAs (spRNAs) play an important regulatory role in bacterial small RNA (sRNA) networks, but their engineering and quantitative systems-level properties are unexplored. Here, we design, build, and quantitatively characterize synthetic spRNA-based gene circuits in E. coli. We establish multiple design strategies for synthetic spRNAs, engineering the first synthetic spRNAs. We show that these synthetic spRNAs can reversibly de-repress sRNA-regulated gene expression, demonstrate tuneable control of gene expression, and extend these designs to multi-target regulation. Through the use of time-resolved continuous-culture characterization in Chi.Bio together with absolute fluorescent protein quantification, we generated a quantitative dynamical dataset for model fitting and mechanistic analysis. Sequential model development showed that recapitulating the observed circuit dynamics required incorporation of Hfq-mediated resource competition, often overlooked in models of sRNA-based synthetic gene circuits. The extended model captured promoter, sRNA, and sponge circuit behavior and was used to investigate quantitative properties of spRNA-mediated regulation, the first such quantitative investigation of spRNA-based regulation. Model-based quantitative investigations further suggest that spRNAs can tune response functions, modulate thresholds and leakiness, alter response times, improve disturbance rejection in some regimes, increase effective specificity, and buffer regulatory output against sRNA mutation. Together, these results establish synthetic spRNAs as a new post-transcriptional tool for bacterial synthetic biology and provide a quantitative framework for understanding natural and engineered spRNA-mediated regulation.
|
|
Scooped by
mhryu@live.com
Today, 12:12 PM
|
Environmental contamination by heavy metals, organic pollutants, and industrial waste poses a pressing global challenge that demands advanced, sustainable remediation technologies. Conventional remediation approaches, including soil excavation and chemical treatment, are costly, generate secondary pollutants, and lack versatility across contamination types. Phytoremediation offers a cost-effective and environmentally safe alternative; however, it is constrained by slow pollutant uptake rates, limited degradation efficiency for persistent organic compounds, and inadequate real-time monitoring capabilities. This review presents a novel interdisciplinary framework that synergistically integrates phytoremediation with nanotechnology, biotechnology, and information technology (IT) to overcome these inherent limitations. Nanotechnology facilitates pollutant detoxification through engineered nanoparticles (NPs) that enhance metal bioavailability and degrade organic pollutants. Biotechnology encompassing genetic engineering and microbial-assisted remediation enhances plant metal accumulation, stress tolerance, and pollutant degradation pathways. IT approaches, including artificial intelligence (AI), remote sensing, and big data analytics, enable real-time monitoring, adaptive strategy optimization, and predictive modelling of remediation outcomes. Together, these technologies constitute a multi-dimensional remediation framework aimed at minimizing environmental damage and promoting ecological sustainability. This review synthesizes recent advancements, highlights key knowledge gaps, and identifies future research directions for next-generation phytoremediation.
|
|
Scooped by
mhryu@live.com
Today, 9:29 AM
|
lthough protein engineering and laboratory evolution have been used to optimize prime editors, we show that previous changes that improve prime editor efficiency also compromise protein stability and expression level, limiting performance. To address these limitations, we apply structure-informed artificial intelligence-guided methods such as the inverse-folding network ProteinMPNN to redesign the reverse transcriptase (RT)domains of engineered and evolved prime editors while preserving regions essential for catalysis. Redesigned RTs are extensively mutated, with 30–163 amino acid substitutions, and exhibit enhanced folding stability and soluble expression and up to twofold higher intracellular prime editor protein levels following mRNA delivery. Redesigned PE8 prime editors demonstrate enhanced editing efficiencies across multiple ex vivo contexts, including in several human primary cell types and via several delivery modalities. In mice, editing efficiency is up to 2.9-fold higher than that of state-of-the-art PE6, PE7 and PEmax prime editors. These findings demonstrate a generalizable approach for augmenting laboratory evolution to improve genome editing agents. A computational redesign strategy improves evolved prime editors.
|
|
Scooped by
mhryu@live.com
Today, 12:13 AM
|
Quantitative analysis of bacterial dynamics in time-lapse microscopy requires robust tracking pipelines, yet selecting and optimizing algorithms for specific experiments remains challenging. Indeed, microbiologists are confronted with numerous algorithms that must be carefully chosen and parameterized to achieve optimal tracking for their experiments. We present an automated methodology to determine optimal tracking configurations for microbiological applications. It is based on TrackMate 8, a novel version of the TrackMate Fiji plugin extended with microbiology-specific tools. Our approach systematically evaluates algorithm-parameter combinations optimizing biologically relevant metrics (e.g., cell-cycle accuracy, bacterial morphology) and includes: (1) integration of deep-learning algorithms (Omnipose, YOLO, Trackastra) adequate for bacterial images in TrackMate; (2) a TrackMate-Helper extension for parameter optimization; and (3) a tracking and segmentation editor for tracking ground-truth generation. We demonstrate the effectiveness of the methodology on two use cases showing its adaptability to diverse experimental conditions. This methodology enables microbiologists with a widely applicable, automated framework to optimize tracking pipelines, facilitating quantitative analysis in bacterial imaging.
|
|
|
Scooped by
mhryu@live.com
Today, 6:08 PM
|
Antimicrobial resistance remains a significant global threat to human health, but microorganisms have long been a crucial source of novel antibiotics. The widely distributed gram-positive bacterium Bacillus subtilis produces an abundance of secondary metabolites, and their antibacterial activities could have significant applications in food, agriculture, and aquaculture areas. These secondary metabolites exert antibacterial effects through mechanisms such as microbial cell membrane structure disruption, cell wall synthesis interference, and cellular metabolic activity inhibition. In contrast to microorganisms such as Streptomyces, B. subtilis forms characteristic biofilms and exhibits quorum sensing, which play important roles in the production of secondary metabolites and their antimicrobial effects. However, limited attention has been focused on the unique molecular mechanisms associated with biofilms and quorum sensing. In this review, we first summarize the typical secondary metabolites produced by B. subtilis. We then mainly focus on the molecular mechanisms associated with the regulation of biofilms and quorum sensing by antimicrobial secondary metabolites, and the effects of biofilms and quorum sensing on the biosynthesis of antimicrobial secondary metabolites. The applications of antimicrobial secondary metabolites in the fields of food, agriculture, and fisheries, based on the regulation of biofilm and quorum sensing, are also summarized. Finally, we highlight the need for further research into the regulatory networks related to biofilms, quorum sensing, and metabolites to facilitate a deeper understanding of the antimicrobial properties of B. subtilis, which may provide theoretical support for the development of novel antimicrobial food technologies.
|
|
Scooped by
mhryu@live.com
Today, 5:57 PM
|
Orfamide A, a lipopeptide produced by Pseudomonas protegens Pf-5, is a key determinant of its biocontrol properties. In this study, we investigated the regulatory interactions among the GacS/A two-component system, small RNAs (sRNAs), repressor proteins, and two LuxR-type transcription factors in orfamide A biosynthesis. We found that GacS/A indirectly regulates orfamide A production by enhancing transcription of three sRNAs (RsmX, RsmY, and RsmZ). RsmY and RsmZ synergistically relieve repression by RsmA and RsmE, while RsmX plays a lesser role, likely counteracting only one repressor. LuxR-type transcription factors, LuxR1 and LuxR2, which positively regulate orfamide A synthesis, are directly repressed by RsmA and RsmE via binding to their 5′ untranslated regions, linking them to the Gac–Rsm signaling cascade. We further demonstrated that LuxR2 activates luxR1 expression, which in turn facilitates orfamide A production by binding to the promoter of the orfamide A biosynthetic gene cluster. Importantly, we showed that this entire regulatory cascade operates in the rhizosphere and directly influence biocontrol efficacy. These findings provide a comprehensive understanding of the Gac–Rsm–LuxR pathway in orfamide A biosynthesis and offer valuable insights for the development of biocontrol agents based on Pseudomonas strains.
|
|
Scooped by
mhryu@live.com
Today, 5:48 PM
|
The rise in antimicrobial resistance underscores the need for innovative strategies to combat gastrointestinal infections. Probiotics such as E. coli Nissle 1917 (EcN) offer promising options, but the molecular mechanisms underlying their protective effects remain unclear. We introduce a G66R point mutation in FimH, creating a high-binding EcN variant that more effectively prevents Salmonella Typhimurium attachment and induces a distinct host transcriptional profile, shifting toward adaptive rather than innate inflammatory signaling. In vivo, EcNG66R pretreatment significantly reduced intestinal colonization, fecal shedding, and systemic spread, and prevented splenic enlargement compared with EcNWT. Protection was associated with a marked expansion of CD4+ and CD8+ T cells, essential for clearing intracellular pathogens. EcNG66R further enhanced “readiness” in the spleen under non-infected conditions, without adverse effects on host physiology. EcNG66R thus functions as a dual-action probiotic—improving competitive exclusion while priming cytotoxic T-cell-mediated protection—and provides a promising platform for developing next-generation microbe-based therapies.
|
|
Scooped by
mhryu@live.com
Today, 5:30 PM
|
The twin-arginine translocation (Tat) system is the only general pathway for the transport of folded proteins across energized biological membranes. It is found in the bacterial or archaeal cytoplasmic membrane, the plant thylakoid membrane or the inner membrane of plant mitochondria. The biological importance of this translocation system can be exemplified by the fact that all bacterial or plant photosynthesis and photosynthetic oxygen evolution on earth requires this system. Despite many biochemical and biophysical studies, the Tat mechanism has been puzzling since the system was discovered in the 1990ies. Important characteristics of the Tat system could not be explained, and also recent high resolution structures of the Tat system’s core with bound substrate has not led to a general transport mechanism yet. In this integrative review, we attempted to answer the key open questions relevant to the Tat mechanism and thereby developed an in its molecular detail new comprehensive explanation of how folded proteins are translocated across membranes by the Tat system.
|
|
Scooped by
mhryu@live.com
Today, 5:23 PM
|
Virus-like particles (VLPs) are widely used as noninfectious platforms for vaccines, drug delivery, and synthetic biology. Here we report a bottom-up approach to generate liposomal VLPs via post-insertion of protein–lipid conjugates into preformed liposomes. Proteins were covalently coupled to a benzylguanine (BG) lipid via the bioorthogonal and irreversible SNAP-tag/BG reaction, and the resulting protein–lipid conjugate enabled detergent-free insertion into liposomal lipid bilayers. Using a SNAP-tagged GFP conjugate, we first confirmed membrane association by liposome cosedimentation and detected increased GFP fluorescence on individual liposomes by flow cytometry. We next applied the method to the SARS-CoV-2 Spike ectodomain. A Spike-SNAP recombinant protein was expressed in Expi293F cells, and its conjugation capability with BG lipid was verified by a binding inhibition assay using a fluorescent BG substrate. Incubation of virus-sized liposomes with Spike–lipid conjugates resulted in lipid-anchor-dependent recovery of Spike in the liposome fraction and increased Spike-associated fluorescence by flow cytometry without detectable changes in particle size. Negative-stain transmission electron microscopy showed a virus-like texture on Spike-modified liposomes compared with unmodified liposomes. In an ACE2 plate-binding assay, Spike-modified liposomes showed higher binding ability than unmodified liposomes, and the binding was reduced by Congo Red, a reported inhibitor of the Spike–ACE2 interaction, and a neutralizing anti-Spike antibody. By decoupling the design of the conjugate from that of the liposome, this post-insertion approach can facilitate rapid and systematic optimization of VLPs for mechanistic investigations and application-oriented studies.
|
|
Scooped by
mhryu@live.com
Today, 4:43 PM
|
Oncolytic bacteria have emerged as a promising platform for targeted cancer therapy owing to their intrinsic ability to preferentially colonize tumor tissues, induce direct tumor cell killing, and remodel the tumor microenvironment to activate antitumor immunity. However, native bacteria alone rarely meet the requirements of precision oncology, particularly in terms of spatial specificity, temporal control, and safety. Recent advances in synthetic biology have enabled the construction of stimulus-responsive gene circuits that confer programmable control over therapeutic gene expression in tumor-colonizing bacteria by coupling defined exogenous triggers or endogenous tumor-associated cues to tightly regulated genetic programs. These engineered systems support the tumor-specific delivery of diverse therapeutic payloads, including cytotoxic agents, cytokines, immunomodulatory ligands, prodrug-converting enzymes, metabolic modulators, and nucleic acid-based therapeutics, while minimizing off-target activity. This review thus summarizes recent developments in stimulus-responsive oncolytic bacteria, highlights key design principles and performance trade-offs, and discusses emerging strategies to advance bacteria as programmable living therapeutics for cancer treatment.
|
|
Scooped by
mhryu@live.com
Today, 3:50 PM
|
While Chlamydomonas is a widely used model organism, nuclear transgene expression in this system is well documented to face notorious difficulties. Here, we show that 5′ untranslated region (UTR) introns can play a positive role in reducing gene silencing. By screening promoter-5′ UTR complexes containing introns (PUTRis) from highly expressed genes, we identify many candidates that outperform the two most widely used promoters, HR and PSAD. Truncation analyses reveal that intron removal increases gene silencing to a similar extent as promoter deletion. These 5′ UTR introns promote open chromatin configurations that support gene expression. Leveraging two identified PUTRis, we achieve robust expression of previously recalcitrant Chlamydomonas proteins and visualize the subcellular localization of over 100 such proteins. Taken together, our findings demonstrate the role of 5′ UTR introns in reducing gene silencing while providing the research community with molecular tools for algal genetic engineering.
|
|
Scooped by
mhryu@live.com
Today, 12:36 PM
|
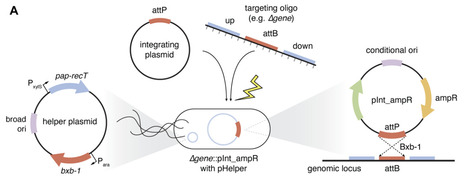
The continual advancement of genetic tools has been critical to our modern understanding of bacteria, with transposons, plasmids, and homologous recombination becoming workhorses of molecular microbiology. However, precisely specified reverse genetic approaches remain painstakingly slow and inaccessible, particularly in non-model strains. This reality is exemplified by the opportunistic pathogen, Pseudomonas aeruginosa (Pa), where conventional allelic exchange remains the dominant reverse genetic method. Here, we adapt a rapid genetic toolkit for use in Pa, relying directly on commercially available oligonucleotides (120 bases) to create precise genomic mutations through homologous recombination (i.e. oligo recombineering). Oligo Recombineering followed by Bxb-1 Integrase Targeting (ORBIT) uses a short attachment site for an integrating plasmid, which provides traditional antibiotic selection and can also carry flexible cargo. We establish Pa ORBIT works effectively for gene deletion without off target mutations, optimize protocol parameters (e.g. oligo length, electroporation), and demonstrate markerless and clean deletions. Importantly, our toolkit works well in clinical Pa strains as demonstrated by constructing efflux pump deletions in three different isolates. To test the high throughput capabilities of Pa ORBIT, we created over 160 degron-based hypomorphs (i.e. knockdowns) across 43 essential proteins in a pooled mutant library. Upon screening this library with and without antibiotics, we identify highly vulnerable essential proteins and hypomorphs that display synergy with clinical drugs. Therefore, ORBIT can be used for cutting edge low and high throughput investigations in this priority pathogen, setting the stage for answering critical basic and clinical science questions.
|
|
Scooped by
mhryu@live.com
Today, 12:26 PM
|
Stability is a desirable property for agricultural microbiomes, but there is a poor understanding of the mechanisms that mediate microbial community stability. A representative bacterial synthetic community from maize roots has been proposed by Niu et al. (2017, PNAS, 114:E2450) as a model system to study microbiome stability. This SynCom assembles stably when all seven members are present, but community diversity collapses without the keystone E. ludwigii strain. In this study, we used complementary in vitro experiments and in silico metabolic modelling to assess the role of metabolites for the stability of this SynCom, by defining the metabolic niches occupied by each strain, as well as their cross-feeding phenotypes and B-vitamin dependencies. We show that the individual member strains occupy complementary metabolic niches, measured by the depletion of distinct metabolites in exometabolomic experiments, as well as contrasting growth phenotypes on diverse carbon substrates, patterns which are largely recapitulated by computational simulations. Minimal medium experiments show that the established seven-member community comprises a mixture of prototrophic and auxotrophic strains. Correspondingly, experimental and in silico cross-feeding phenotypes showed that spent media harvested from the prototrophic strains can sustain growth of two auxotrophs and let to the identification of B-vitamin dependencies. Altogether, this study highlights the complementary power of in vitro and in silico approaches and suggests that the metabolic mechanisms of this SynCom can serve as design principles to inform the rational assembly of stable plant-associated microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:16 PM
|
Extracellular pH is a key microenvironmental factor shaping cell physiology and disease, creating a need for quantitative biosensors that can capture dynamic changes in pHe at the surface of individual living cells. Here, we develop a genetically encoded, ratiometric extracellular pH biosensor through systematic screening of a modular library of membrane-display designs that combine SEpHluorin with a pH-stable reference fluorophore. Screening identified a cell-surface-localised mKate2-SEpHluorin construct, named SurpHer, that exhibits dynamic ratiometric responses across the pHe range of 6 - 7.8. SurpHer shows robust membrane localization and extracellular pH responsiveness across diverse human cell types including HEK293T, PANC-1 and MDA-MB231 cells. Following stable integration in MDA-MB-231 cells, SurpHer enabled time-course imaging of pHe gradients in a microfluidic platform for modelling tumor microenvironments. SurpHer enables real-time interrogation of the pericellular pH environment of tumor cells and, more broadly, provides a strategy to probe microenvironmental pH dynamics across diverse biological contexts.
|
|
Scooped by
mhryu@live.com
Today, 12:04 PM
|
Accurate monitoring of canopy nitrogen content is essential for sustainable nitrogen management, yield improvement, and environmental protection in industrial maize production. However, the high dimensionality of hyperspectral data and the limited accuracy and interpretability of existing models hinder practical applications. This study was conducted in Heilongjiang Province, China, using the maize cultivar Jinboshi. Genetic Algorithm (GA), Successive Projections Algorithm (SPA), and their hybrid strategy were compared for spectral band optimization. Sensitive vegetation indices were selected using multiple evaluation criteria, and a 0–2 order fractional-order derivative (FOD) method was applied to construct optimal two-dimensional (2D) and three-dimensional (3D) spectral indices. A stacked ensemble learning model was developed using XGBoost, GBDT, and Ridge as base learners and Bayesian Ridge as the meta-learner. Interpretability techniques were applied to analyze feature contributions. The GA–SPA hybrid strategy effectively improved key spectral band selection. The 3D spectral index based on FOD achieved superior performance compared to vegetation indices and 2D indices (R2p = 0.801, RMSEP = 0.481). The optimized multi-source feature set combined with the stacked ensemble model yielded the best performance (R2p = 0.826, RMSEP = 0.450). Features from the red-edge and near-infrared regions, along with the 3D index, were the primary contributors to model predictions, consistent with plant nitrogen physiology. The proposed framework, integrating feature optimization, advanced modeling, and interpretability analysis, provides an effective tool for precise nitrogen management in industrial maize and supports improved production efficiency with reduced environmental impact.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Language models trained on biological sequences are advancing inference tasks from the scale of single proteins to that of genomic neighborhoods. Here, we introduce ProteomeLM, a transformer-based language model that uniquely operates on entire proteomes from species spanning the tree of life. ProteomeLM is trained to reconstruct masked protein embeddings using the whole proteomic context, yielding contextualized protein representations that reflect proteome-scale functional constraints. Notably, ProteomeLM’s attention coefficients encode protein–protein interactions (PPI), despite being trained without interaction labels. Furthermore, it enables interactome-wide PPI screening that is substantially more accurate, and orders of magnitude faster, than amino acid coevolution-based methods. We further develop ProteomeLM-PPI, a supervised model that combines ProteomeLM embeddings and attention coefficients to achieve state-of-the-art PPI prediction across benchmarks and species. Finally, we introduce ProteomeLM-Ess, a supervised gene essentiality predictor that generalizes across diverse taxa. Our results demonstrate the potential of proteome-scale language models for addressing function and interactions at the organism level.
|