Your new post is loading...

|
Scooped by
?
Today, 12:55 PM
|
Multicellularity has evolved dozens of times in many branches across the Tree of Life, in each case producing a new kind of individual that has the potential for division of labor among its constituent cells. In many of these instances, nascent multicellularity has remained facultative, manifesting only under conditions where it provides a decisive fitness advantage to the genome in which it has arisen. Here we investigate the mechanistic basis for, and the selective advantages of, cryptic multicellularity in wild strains of the yeast Saccharomyces cerevisiae. Meiotic purification of this trait, followed by genome sequencing, indicates that the genome of diploid champagne yeast DB146 is homozygous at the AMN1 locus (Antagonist of Mitotic exit Network) for an allele that dysregulates post-mitotic cell separation. Expression of this trait in haploid derivatives of DB146 results in clonal multicellular clusters in which daughters remain attached to mother cell walls. Systematic analysis of viability in haploid and diploid DB146 derivatives subjected to benign conditions as well as to starvation, desiccation, and low temperature at different cell titers demonstrates that haploid multicellular variants exhibit higher survivorship under conditions that wild yeast likely experience when they overwinter. Examining a collection of other wild strains, we find that ~20% exhibit ploidy-dependent expression of clonal multicellularity. Altogether, our data suggest that in yeast that sporulate when starved, cryptic multicellularity may come under balancing selection because haploid multicellular progeny enjoy a transient survival advantage that fades once favorable growth conditions resume.
|
|
Scooped by
?
Today, 12:40 PM
|
RNApdbee 3.0 https://rnapdbee.cs.put.poznan.pl/ offers an advanced pipeline for comprehensive RNA structural annotation, integrating 2D and 3D data to build detailed nucleotide interaction networks. It classifies base pairs as canonical or noncanonical using the Leontis-Westhof and Saenger schemes and identifies stacking, base-ribose, base-phosphate, and base-triple interactions. The tool handles incomplete or modified residues, marking missing nucleotides and distinguishing noncanonical base pairs for accurate and effective visualization. Results are provided in standard formats - namely, extended dot-bracket notation, BPSEQ, and CT - and in highly valuable graphical visualizations. RNApdbee decomposes secondary structures into stems, loops, and single strands and offers flexible pseudoknot encoding. Its unified framework addresses inconsistencies across structural data formats by standardizing all inputs to PDBx/mmCIF and integrating seven widely used annotation tools. Finally, RNApdbee ensures reliable, format-independent, and comprehensive RNA structural annotation and interpretation.
|
|
Scooped by
?
Today, 12:32 PM
|
The genome folds inside the cell nucleus into hierarchical architectural features, such as chromatin loops and domains. If and how this genome organization influences the regulation of gene expression remains only partially understood. The structure-function relationship of genomes has traditionally been probed by population-wide measurements after mutation of critical DNA elements or by perturbation of chromatin-associated proteins. To circumvent possible pleiotropic effects of such approaches, we have developed OptoLoop, an optogenetic system that allows direct manipulation of chromatin contacts by light in a controlled fashion. OptoLoop is based on the fusion between a nuclease-dead SpCas9 protein and the light-inducible oligomerizing protein CRY2. We demonstrate that OptoLoop can drive the induction of contacts between genomically distant, repetitive DNA loci. As a proof-of-principle application of OptoLoop, we probed the functional role of DNA looping in the regulation of the human telomerase gene TERT by long-range contacts with the telomere. By analyzing the extent of chromatin looping and nascent RNA production at individual alleles, we find evidence for looping-mediated repression of TERT. In sum, OptoLoop represents a novel means for the interrogation of structure-function relationships in the genome at single-allele resolution.
|
|
Scooped by
?
Today, 11:36 AM
|
Novel approaches to genome engineering are crucial to rapidly advance the capabilities of strain engineering and synthetic biology. With ongoing developments in DNA editing techniques, researchers have begun to engineer organisms at higher throughput and can now perform multiple genome modifications simultaneously. As laboratory automation becomes more accessible, workflows are being transferred to robot-assisted platforms, enabling large-scale and highly parallelized genome editing campaigns. These platforms play a key role in fully utilizing the potential of modern molecular biology tools. Here, we review recent developments in technologies for high-throughput, multiplexed, and automated strain engineering in prokaryotic and eukaryotic organisms.
|
|
Scooped by
?
November 7, 5:11 PM
|
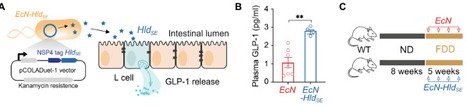
Western diets reduced in fiber promote dysbiosis and exacerbate colitis, with short-chain fatty acids (SCFAs) from fiber fermentation known to regulate glucagon-like peptide 1 (GLP-1) secretion. Whether GLP-1 dysregulation directly links diet-induced dysbiosis to colitis severity, and if this pathway can be therapeutically targeted independently of dietary fiber, remains unclear. Here, we show that dextran sulfate sodium (DSS)–induced colitis severity correlates with compensatory GLP-1 increases, while receptor blockade worsens damage, confirming GLP-1’s protective role during colitis. Fiber deficiency impaired L cell function and GLP-1 release, increasing colitis susceptibility. GLP-1 receptor agonist reversed these effects, restoring barrier integrity and accelerating recovery. We engineered a probiotic, releasing a microbial peptide, that locally elevates GLP-1, which normalized gut parameters in fiber-deprived mice and alleviated colitis via GLP-1–dependent mechanisms, including improved metabolism, antimicrobial defenses, and barrier restoration. Our findings mechanistically connect fiber deficiency to colitis through GLP-1 and demonstrate that probiotic-mediated GLP-1 modulation can bypass dietary fiber requirements to maintain gut homeostasis.
|
|
Scooped by
?
November 7, 5:05 PM
|
The exploration of post-translational modifications (PTM) within the proteome is pivotal for advancing our understanding of disease and the function of cancer therapeutics. However, identifying genuine sites of PTMs introduced or removed by an enzyme of interest amid numerous candidates is challenging. We present a machine learning (ML)-driven search method, which combines ML with enzyme-mediated modification of complex peptide arrays to predict unexplored PTM sites for an enzyme of interest. Experimental validation confirmed that this approach correctly predicted 37-43% of proposed PTM sites, unveiling candidate sites of the methyltransferase SET8 and the deacetylases SIRT1-7. Our approach marks an important performance increase over traditional in vitro methods across separate enzyme classes. Mass spectrometry analysis confirmed the dynamic methylation status of several predicted SET8 substrates, and the deacetylation of 64 unique sites identified for SIRT2. This method has also revealed changes in SET8-regulated substrate network among breast cancer missense mutations, collectively revealing insight into differential enzyme function in disease. By disentangling the substrate features that dictate PTM-inducing enzyme specificity, this approach demonstrates potential in uncovering enzyme-substrate networks within PTM pathways. Understanding post-translational modifications (PTMs) is crucial for advancing disease research and cancer therapeutics, yet identifying specific enzyme-induced PTM sites remains challenging. Here, the authors introduce a machine learning-driven method combined with high-throughput peptide array synthesis, significantly enhancing PTM site prediction accuracy and revealing insights into enzyme-substrate networks, with implications for cancer research.
|
|
Scooped by
?
November 7, 4:57 PM
|
Understanding microbial interactions is fundamental for exploring population dynamics, particularly in microbial communities where interactions affect stability and host health. Generalized Lotka-Volterra (gLV) models have been widely used to investigate system dynamics but depend on absolute abundance data, which are often unavailable in microbiome studies. To address this limitation, we introduce an iterative Lotka-Volterra (iLV) model, a novel framework tailored for compositional data that leverages relative abundances and iterative refinements for parameter estimation. The iLV model features two key innovations: an adaptation of the gLV framework to compositional constraints and an iterative optimization strategy combining linear approximations with nonlinear refinements to enhance parameter estimation accuracy. Using simulations and real-world datasets, we demonstrate that iLV surpasses existing methodologies, such as the compositional LV (cLV) and the generalized LV (gLV) model, in recovering interaction coefficients and predicting species trajectories under varying noise levels and temporal resolutions. Applications to the lynx-hare predator-prey, Stylonychia pustula-P. caudatum mixed culture, and cheese microbial systems revealed consistency between predicted and observed relative abundances showcasing its accuracy and robustness. In summary, the iLV model bridges theoretical gLV models and practical compositional data analysis, offering a robust framework to infer microbial interactions and predict community dynamics using relative abundance data, with significant potential for advancing microbial research.
|
|
Scooped by
?
November 7, 4:46 PM
|
Untargeted tandem mass spectrometry (MS/MS)-based metabolomics enables broad characterization of small molecules in complex samples, yet the majority of spectra in a typical experiment remain unannotated, limiting biological interpretation. Reference data-driven (RDD) metabolomics addresses this gap by contextualizing spectra through comparison to curated, metadata-annotated reference datasets, allowing inference of spectrum origins without requiring exact structural identification. Here, we present an open-source RDD metabolomics platform comprising a user-friendly web application and a Python software package that perform RDD analyses directly from molecular networking outputs generated by GNPS. The tools support visualization and statistical analysis of RDD results, including interactive bar plots, heat maps, principal component analysis, and Sankey diagrams. We illustrate the approach using a hierarchical reference dataset of 3,500 food items to derive dietary patterns from stool metabolomics data of omnivore and vegan participants. The analysis reveals clear dietary group separation, demonstrating how RDD metabolomics can extract biologically meaningful patterns from otherwise unannotated spectra. Thus, the RDD metabolomics platform removes technical barriers for the metabolomics community to adopting reference data-driven analysis, with the functionality freely available at https://github.com/bittremieuxlab/gnps-rdd and https://gnps-rdd.streamlit.app/.
|
|
Scooped by
?
November 7, 4:34 PM
|
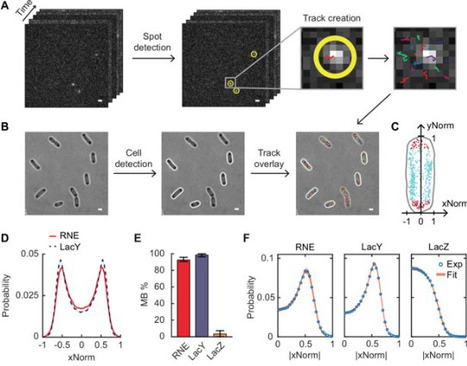
In Escherichia coli, RNase E, a central enzyme in RNA processing and mRNA degradation, contains a catalytic N-terminal domain, a membrane-targeting sequence (MTS), and a C-terminal domain (CTD). We investigated how MTS and CTD influence RNase E localization, diffusion, and function. Super-resolution microscopy revealed that ~93% of RNase E localizes to the inner membrane and exhibits slow diffusion similar to polysomes. Comparing the native amphipathic MTS with a transmembrane motif showed that the MTS confers slower diffusion and stronger membrane binding. The CTD further slows diffusion by increasing mass but unexpectedly weakens membrane association. RNase E mutants with partial cytoplasmic localization displayed enhanced co-transcriptional degradation of lacZ mRNA. These findings indicate that variations in the MTS and the presence of the CTD shape the spatiotemporal organization of RNA processing in bacterial cells, providing mechanistic insight into how RNase E domain architecture influences its cellular function.
|
|
Scooped by
?
November 7, 4:21 PM
|
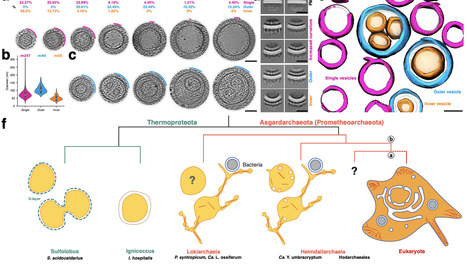
The emergence of eukaryotes from a merger between an archaeon and a bacterial cell ~two billion years ago involved a profound change in cellular organization. While the order in which different features of the eukaryotic cell arose remains a matter of controversy, close archaeal relatives of eukaryotes have recently been identified that possess homologues of eukaryotic trafficking machinery and a complex cell architecture. The members of this phylum, the Asgard archaea (syn. Prometheoarchaeota) described so far, however, lack internal membrane-bound compartments, and therefore have shed little light on origins of the hallmark eukaryotic endomembrane system. Here we report the cell biological analysis of a member of the Heimdallarchaeia, Candidatus "Yibarchaeum umbracryptum" in enriched mixed microbial communities. Possessing a small genome encoding few homologues of eukaryotic membrane remodelling machinery, Ca. Y. umbracryptum cells in late-stage cultures resemble previously described Asgard archaea with extensive cellular protrusions. Surprisingly, during early stages of culture growth Ca. Y. umbracryptum cells have fewer protrusions but possess numerous intracellular vesicles, most of which have a luminal surface that morphologically resembles the outer coat of the plasma membrane. These data alter our view of eukaryogenesis by identifying a close archaeal relative of eukaryotes with a regulated endomembrane system.
|
|
Scooped by
?
November 7, 3:09 PM
|
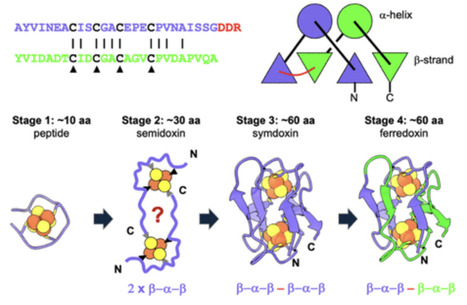
Electron transfer coupled to redox chemistry is at the heart of metabolism. The proteins responsible for moving electrons (protein electron carriers) must have emerged at the origin of life. The small iron–sulfur-binding bacterial ferredoxins were likely among these first proteins. Embedded within the ferredoxin sequence and structure is a symmetry that points to an ancient gene duplication event. Little is understood about the nature of ferredoxins prior to this duplication event or what environmental factors may have driven the selection for more complex forms. The deep-time molecular history of ferredoxins goes back billions of years and cannot be reconstructed by phylogenetic analyses based on amino acid sequences. Here, we use structure-guided protein design to model a fossil half-ferredoxin stage in the evolution of this fold, the semidoxins, and their symmetric full-length counterparts, the symdoxins. Semidoxin designs homodimerize, exhibiting structural, thermodynamic, and electrochemical behaviors in most cases identical to cognate symdoxins. However, the semi- and symdoxin fossil stages behave differently when incorporated into an in vivo electron transfer complementation assay. Both can support bacterial growth dependent on protein expression. Growth rates of bacteria expressing the semidoxins are much more sensitive to oxygen than those of bacteria expressing symdoxins. Motivated by the in vivo functionality of designed semidoxins, we identified putative naturally occurring semidoxins in extant anaerobic microorganisms. This is consistent with the observed in vivo oxygen sensitivity of the semidoxin designs. One natural semidoxin is shown to be folded and redox active. However, it exists as a mixture of monomers and dimers, suggesting a potential connection between semidoxins and even simpler single iron–sulfur cluster-binding peptides.
|
|
Scooped by
?
November 7, 2:21 PM
|
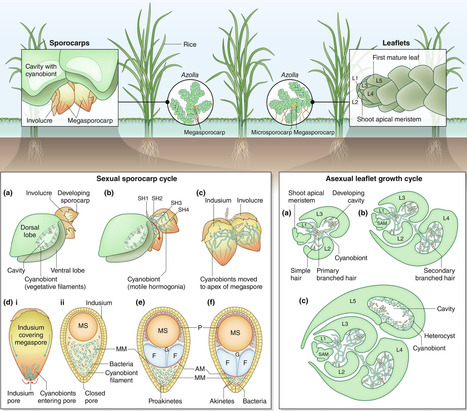
Heritable symbioses exist across eukaryotes with different degrees of intimacy. In most cases, the symbionts are obligate and require inheritance for their survival. On the host side, symbiont retention can facilitate fitness benefits. Only rarely are these symbioses interwoven to the point that host survival relies on the symbiont. In land plants, the symbiosis of the water fern Azolla with its symbiotic cyanobacterium shows such a degree of high co-dependence. The symbiosis originated in the last common ancestor of Azolla and exists continuously for at least 60 million years with no evolutionarily stable, secondary loss of the symbiont reported. This is a feat achieved by interactions on an organellar-like level or those considered recent organelle acquisitions. Yet, Azolla's symbiont is extracellular. How can loss of autonomy concomitant with full co-dependence be accommodated in this extracellular symbiosis? Here, we synthesize what we know from the Azolla symbiosis on the consequences of evolutionary co-dependence and stable symbiont retention. We discuss the need for symbiotic integration into environmental responsiveness if host survival depends on symbiont well-being. Cross-organismal integration of environmental stress responses may be one of the key steps that favor this evolutionarily stable permanent integration.
|
|
Scooped by
?
November 7, 1:56 PM
|
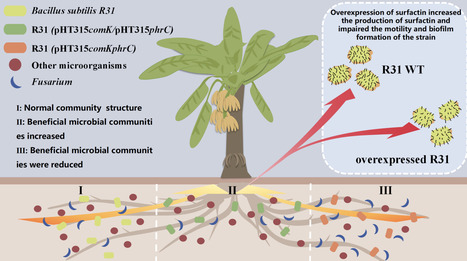
Surfactin, a lipopeptide antibiotic and quorum-sensing (QS) mediator from Bacillus subtilis, has dual functions in microbial ecology and plant disease suppression. This study engineered B. subtilis R31 to overproduce comK and phrC, key regulators of surfactin biosynthesis, increasing surfactin yield by 45% compared to the WT strain. While elevated surfactin enhanced antimicrobial potential, comK-mediated overproduction impaired biofilm formation and swarming motility, but rhizosphere colonization was mostly unaffected. 16S rRNA sequencing of banana rhizospheres showed that surfactin selectively shaped the microbial community by enriching beneficial Bacillus species. Mechanistic studies confirmed surfactin's dual role as an antimicrobial and an intercellular signalling molecule for coordinated development in Bacillus populations. These results reveal the molecular mechanisms of R31-mediated suppression of banana Fusarium wilt and offer a strategy for engineering synthetic microbial consortia by manipulating metabolic signalling pathways.
|
|
|
Scooped by
?
Today, 12:51 PM
|
Complex microbial ecosystems harbor extensive intra-species diversity, but the fitness consequences of this genetic variation are poorly understood in community settings. Here we address this question by competing in vitro gut communities derived from different human donors, revealing the emergent fitness differences between conspecific strains as they competed within larger communities. Most pairs of strains experienced strong and context-dependent selection, even when their parent communities were originally selected in the same nutrient environment. However, these fitness differences typically attenuated over time due to biotic interactions within the community, leading to extended coexistence within many species, and competitive exclusion in others. These results support the view that conspecific strains can fulfill distinct ecological roles when competing within a diverse community, even when their genomic diversity exhibits the hallmarks of a single biological species.
|
|
Scooped by
?
Today, 12:35 PM
|
Metatranscriptomic (MetaT) sequencing provides critical insights into the gene expression and functional activity of microbial communities. However, its utility is limited by the overwhelming abundance of ribosomal RNA (rRNA), which typically represents ≥90% of total RNA. A major obstacle to efficient MetaT analysis is the removal of highly abundant rRNA transcripts present in complex microbial communities, which may contain thousands of species. Although commercial rRNA depletion kits can effectively reduce rRNA content, they are typically optimized for specific host microbiomes and often underperform in others. For example, probes designed for the human gut microbiome frequently show reduced efficiency when applied to non-human samples such as mouse cecal donor samples - a common model in microbiome research. Regardless of the depletion strategy used, designing rRNA removal probes solely based on a microbiome's taxonomic composition often requires an extensive number of probes, making the approach expensive, difficult to manufacture, and sometimes technically impractical. Here, we present RiboZAP, a species-agnostic computational pipeline for designing custom RNase H depletion probes directly from MetaT sequencing data and without prior knowledge of sample composition. Our results show that the probes generated with RiboZAP are efficient for removal of rRNA content, increasing messenger RNA (mRNA), and improving transcriptome coverage. This provides a cost-effective approach to maximize the value of MetaT sequencing.
|
|
Scooped by
?
Today, 12:00 PM
|
Protein design has been transformed by deep generative models ,but at the cost of interpretability, accessibility, and integration with sparse experimental feedback. Here, we introduce Genomic Perception Fusion (GPF), a biologically inspired algorithm that treats DNA not as inert code, but as a linear signal awaiting perceptual reconstruction. GPF transforms nucleotide sequences, augmented with non-coding regulatory context, into a high-order functional representation that predicts stability, solubility, and expression. Built from physicochemical first principles and literature-derived parameters, GPF runs on a laptop in under a second, yet accurately forecasts the effects of surface mutations in green fluorescent protein (GFP). Validated against computational benchmarks, GPF offers a frugal, transparent alternative to black-box design for rapid protein engineering.
|
|
Scooped by
?
Today, 11:33 AM
|
Biofoundries are transforming synthetic biology, with the DNA assembly workflow being critical for successful biofoundry operation. This review examines recent advances in automated DNA assembly strategies for biofoundry, focusing on three key perspectives. First, we discuss emerging platforms ranging from high-throughput and highly efficient systems to affordable and accessible solutions. Second, we explore how standardized design tools enable seamless interoperability across diverse biofoundries, facilitating protocol sharing and reproducibility. Third, we analyze the integration of machine learning into assembly workflows, in which AI-driven systems dynamically optimize protocols, diagnose failures, and close the DBTL loop through real-time learning. These convergent advances are establishing a new paradigm in which experiments continuously improve through iteration, promising to accelerate both fundamental research and industrial applications.
|
|
Scooped by
?
November 7, 5:08 PM
|
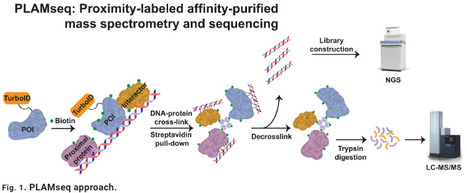
Chromatin immunoprecipitation and coimmunoprecipitation assays are common approaches to characterize the genomic localization and protein interactors, respectively, for a protein of interest. However, these approaches require the use of specific antibodies, which often face sensitivity and specificity issues. On the basis of TurboID, we developed proximity-labeled affinity-purified mass spectrometry and sequencing (PLAMseq), which enables, in the same workflow, identification of the genomic loci and the interacting proteome of a protein of interest. Moreover, PLAMseq can also be applied to specifically map protein interactions and ubiquitin(-like)–modified proteins. We validated PLAMseq with two well-characterized proteins, RNA polymerase II, and CTCF, with excellent robustness and reproducibility. Next, we applied PLAMseq to characterize histone H1 SUMOylation, in which study has remained elusive due to the lack of specific reagents, and found that SETDB1 binds to SUMOylated histones H1.2 and H1.4 that also colocalize with H3K9me3 at repetitive regions of the genome.
|
|
Scooped by
?
November 7, 5:01 PM
|
Protein solubility is a critical physicochemical property influencing protein stability, therapeutic efficacy, and overall developability in drug discovery. However, traditional experimental methods for assessing solubility are often resource-intensive and time-consuming. To address these limitations, computational approaches leveraging artificial intelligence have emerged, yet current models generally treat qualitative classification and quantitative regression as separate tasks and rely predominantly on sequence-based information, neglecting crucial structural and surface characteristics. Here, we introduce Pro4S, a novel multimodal predictive model that integrates protein language models, structural data, and surface descriptors using advanced contrastive learning techniques. Our unified framework achieves significant improvements in prediction accuracy, robustness, and generalizability for both qualitative and quantitative solubility assessments. Benchmark comparisons demonstrate that Pro4S consistently outperforms existing state-of-the-art predictors across diverse datasets. Furthermore, by applying Pro4S to the emerging area of de novo protein design, we validated a strong correlation between predicted solubility and experimental expression levels, reducing the proportion of non-expressed proteins by 52.7% while retaining 96.7% of highly expressed proteins. This highlights Pro4S's potential to serve as a reliable upfront screening tool for increasing expression success rates and accelerating rational protein engineering.
|
|
Scooped by
?
November 7, 4:53 PM
|
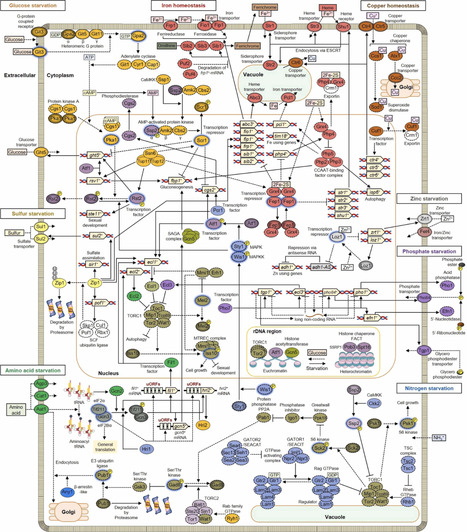
In nature, nutrient-poor environments are more common than exposure to nutrient-rich environments, and living organisms have developed countermeasures to survive nutrient starvation. Increasing research has revealed beneficial aspects of starvation for an individual’s life, including lifespan extension. The fission yeast Schizosaccharomyces pombe is a model unicellular eukaryotic organism and has greatly contributed to the understanding of various cellular processes, including the cell cycle, cell morphology, sexual development, cell lifespan, and nutritional responses. Traditionally, research on starvation in fission yeast has focused on glucose starvation and nitrogen starvation. Recently, studies on cellular responses to the starvation of various nutrients, such as phosphorus, sulfur, iron, zinc, copper, and amino acids have been reported, revealing similarities and differences among the various types of nutrient starvation. In fission yeast, Ecl proteins, which are conserved among fungi, can sense the starvation of multiple nutrients. These proteins also repress the target of rapamycin complex 1 (TORC1), which is conserved across eukaryotes. They channel a variety of starvation signals into common cellular responses, such as growth arrest, sexual differentiation, autophagy, and lifespan extension. This review summarizes and discusses the signaling mechanisms involved in the initial cellular responses of fission yeast to the starvation of various nutrients.
|
|
Scooped by
?
November 7, 4:39 PM
|
The development of hydrogenases capable of operating under oxygenic photosynthetic conditions remains a key challenge for sustainable biohydrogen production. In this study, we developed a series of chimeric NAD+-reducing [NiFe]-hydrogenases (SH) combining structural elements from the O2-tolerant SH of Cupriavidus necator (CnSH) and the ferredoxin-interacting SH of Synechocystis sp. PCC6803 (SynSH). By engineering chimeric HoxU and HoxF subunits, we developed constructs─MixSH, Ch-HoxEFSyn+UCn, and Ch-HoxUswapCTD─that successfully couple the CnHoxYH hydrogenase module to the SynHoxEFU reductase module while retaining O2 tolerance and enhancing interaction with reduced ferredoxin. The lithoautotrophic growth of C. necator confirmed the tolerance of these variants to O2, while activity assays in Synechocystis demonstrated partial hydrogenase function, including H2 consumption and fermentative H2 production. Notably, Ch-HoxEFSyn+UCn retained ferredoxin interaction despite lacking the [4Fe4S]U4 cluster, showing [2Fe2S]F2 in HoxF as a functional ferredoxin-binding site. Moreover, we achieved the artificial integration of a [2Fe2S] cluster into CnHoxF and identified the CnHoxF N-terminal domain as structurally and functionally analogous to SynHoxE. Although electron transfer efficiency and activity in Synechocystis remained limited, this work validates the modular engineering of [NiFe]-hydrogenases, uniting O2-tolerance with ferredoxin interaction and offering a foundational step toward photosynthesis-coupled H2 production.
|
|
Scooped by
?
November 7, 4:30 PM
|
Multiple global change drivers have caused a large carbon (C) debt in our soils. To remedy this debt, understanding the role of microorganisms in soil C cycling is crucial to tackle the C soil loss. Microbial carbon use efficiency (CUE) is a parameter that captures the formation of microbially-derived soil organic matter (SOM). While it is known that biotic and abiotic drivers influence CUE, it remains unclear whether bacteria, fungi and their interactions influence the formation of microbially-derived SOC and its persistence in soils. Here, we combined the inoculation of distinct communities (a biotic factor) grown at different moisture levels (an abiotic factor) to manipulate the formation of microbial necromass in model soil. In a follow-up experiment, we then evaluated the persistence of this previously formed microbially-derived C to decomposition. While we show that necromass formation reflects the microbial community composition, the SOC formed within the most complex community of bacteria and fungi seems to be more resistant to decomposition compared to the SOC formed within the simpler communities (bacteria and fungi simple community, bacteria only and fungi only communities). Moreover, fungal necromass proved to be more thermally-stable than bacterial necromass, if this necromass is formed with both bacteria and fungi present. Our findings reveal that although abiotic factors can influence microbial physiology, the biological origin of microbially-derived C and the co-occurrence of fungal and bacterial growth were the stronger drivers explaining SOM persistence in these soils, suggesting the importance of microbial succession in SOC stabilization.
|
|
Scooped by
?
November 7, 3:13 PM
|
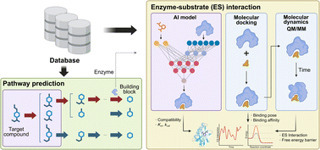
Natural products are a major source of bioactive compounds, yet elucidating their biosynthetic pathways remains a major challenge due to complex genotype–phenotype relationships. Recent advances in computational approaches, particularly artificial intelligence (AI) and mechanistic modeling, are transforming this field. This highlight examines key databases that underpin computational studies, AI-driven methods for predicting biosynthetic pathways and enzyme–substrate interactions, and mechanistic simulations that provide energetic and structural insights. We also discuss current challenges and future opportunities for integrating these strategies to accelerate discovery, engineering, and application of natural products in drug discovery, biotechnology, and synthetic biology.
|
|
Scooped by
?
November 7, 2:22 PM
|
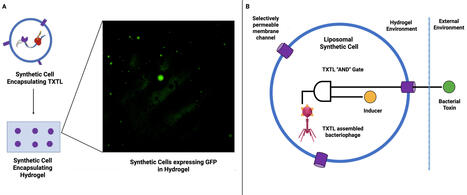
A synthetic cell is a membrane-bound vesicle that encapsulates cell-free transcription/translation (TXTL) systems. It represents a transformative platform for advancing bacteriophage therapy. Building on experimental work that demonstrates (i) modular genome assembly, (ii) high-yield phage TXTL systems, and (iii) smart hydrogel encapsulation, we explore how synthetic cells can address major limitations in phage therapy. The promising advances include point-of-care phage manufacturing, logic-responsive antimicrobial biomaterials, and new chassis to dissect the dynamics of phage-host interactions. We also propose a roadmap for the deployment of synthetic cells as programmable and evolvable tools in the context of laboratory research and translational clinical adoption. droplet
|
|
Scooped by
?
November 7, 2:01 PM
|
This work describes the development of a CRISPR interference (CRISPRi) system for targeted gene repression in bifidobacteria. We first validated the CRISPRi-based approach using Bifidobacterium breve strains engineered to express nuclease-dead orthologs of Cas9 and demonstrated that the CRISPR-Cas system from Streptococcus thermophilus is efficient at targeting both reporter and endogenous genes through the use of single guide RNAs corresponding to the gene of interest. We also developed a one-plasmid system for targeted gene repression in bifidobacteria and demonstrated its utility by targeting genes involved in nucleotide metabolism and carbohydrate metabolism in several species of bifidobacteria. Efficient gene repression was achieved across all tested bifidobacterial species without the requirement for extensive optimization of transformation parameters or sequence optimization to avoid restriction modification systems thus removing the key barriers to genetic manipulation in this genus. This CRISPRi system provides a novel approach to functional genomics in bifidobacteria which facilitates future mechanistic studies in these commercially important microbes.
|