 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:37 AM
|
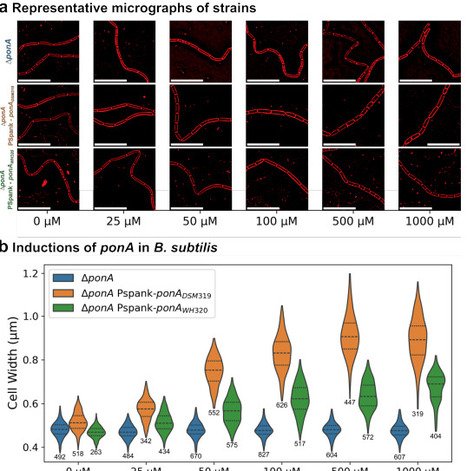
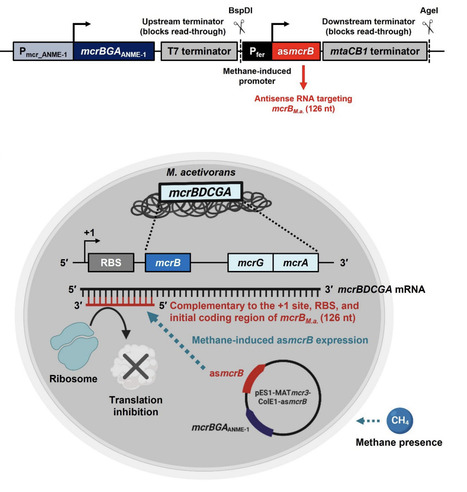
Utilizing methane and carbon dioxide before it can enter the upper atmosphere is beneficial for mitigating climate change as well as for producing valuable chemicals. Because anaerobic methanotrophic archaea (ANME) have not yet been cultured in isolation, we previously reversed methanogenesis by cloning the genes encoding methyl-coenzyme M reductase (Mcr) derived from Black Sea ANME-1 into the methanogen Methanosarcina acetivorans. The resulting engineered archaeal strain captures, rather than produces, methane and may be used to convert methane and carbon dioxide into electricity, acetate, L-lactate, and ethanol. However, the engineered M. acetivorans strain also contains a chromosomal locus encoding its native Mcr (McrM.a.), which produces methane from substrates such as methanol, whereas the heterologously expressed ANME-1 Mcr (McrANME-1) promotes methane oxidation. Therefore, we reasoned that McrM.a. may compete with McrANME-1-mediated reversal of methanogenesis. To enhance the reversal of methanogenesis, here we implemented an antisense RNA (asRNA) silencing approach to suppress McrM.a. during growth on methane while still allowing its expression during routine growth on methanol. We found that silencing McrM.a. during McrANME-1-mediated growth on methane increased ethanol and acetate production by more than an order of magnitude. These results were corroborated by both a more than 10-fold increase in methane utilization by McrANME-1 and a greater than 1,000-fold reduction in the McrM.a. mcrBGA transcript levels under methane-grown conditions. Therefore, asRNA-mediated silencing may be used to enhance methane capture by suppressing production of the host McrM.a. for biotechnological applications.
|
|
Scooped by
mhryu@live.com
Today, 12:10 AM
|
Mushroom-forming Agaricomycete fungi underpin global nutrient cycling and carbon sequestration, and support large and growing markets across food, medicinal supplements, and biomaterials. Yet most commercial and research uses still rely on wild-type strains, highlighting the opportunity for genetic engineering to expand possibilities for both fundamental research and biotechnological applications. In this review, we highlight progress toward synthetic biology in Agaricomycetes, and outline the main barriers that limit predictable genetic engineering. We emphasize engineering constraints unique to mushroom biology, including complex sexual cycles, heterokaryosis, and strain instability during transformation and outgrowth. We then transition to gene expression bottlenecks: the scarcity of characterized promoters and terminators, the challenges for gene integration posed by the condensed nature of Agaricomycete genomes, and the effects of introns and specific sequence motifs. Finally, drawing inspiration from progress in related fungi and other eukaryotes, we highlight the priorities for the field: systematic cross-species evaluation of genetic parts, development of more sophisticated gene-editing strategies, higher-throughput screening methods, and the establishment of a unifying model system. These advances would enable new possibilities in the study and use of Agaricomycetes, establishing these elusive organisms as programmable platforms for sustainable biomanufacturing, designer biomaterials, climate solutions, and mechanistic studies of fungal biology.
|
|
Scooped by
mhryu@live.com
June 4, 11:48 PM
|
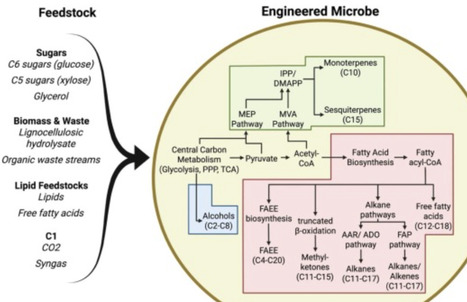
Sustainable aviation fuels (SAF) are critical for decarbonising the hard-to-abate aviation sector, which significantly contributes to global CO2 emissions. Conventional SAF production routes, such as Hydroprocessed Esters and Fatty Acids, Fischer-Tropsch and Alcohol-to-Jet, offer drop-in compatibility but are constrained by feedstock availability, high costs and environmental impacts. This review highlights, as promising alternatives, microbial bioproduction via precision fermentation of SAF-relevant compounds from low-cost feedstocks, with reduced land use and enhanced circularity. Here, we focus on microbially derived SAF precursors such as alcohols, terpenes, fatty acid ethyl esters, methyl ketones and saturated hydrocarbons, as well as recent advances in host engineering, pathway design, and bioprocess optimization.
|
|
Scooped by
mhryu@live.com
June 4, 11:27 PM
|
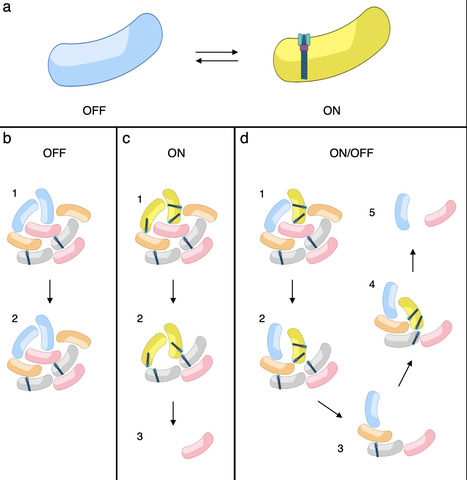
The type VI secretion system (T6SS) is a widespread nanoweapon deployed by bacteria to eliminate competitors in polymicrobial environments, allowing niche colonization or host invasion. Fluorescent microscopy recordings have shown that T6SS expression and/or activation is heterogeneous in clonal populations of many bacterial species. However, it is still unknown whether T6SS heterogeneity is genetically controlled or arises from stochastic processes and what its physiological relevance is. Here, we report that enteroaggregative E. coli (EAEC) exhibits stable phenotypic heterogeneity in T6SS expression. Under iron-limiting conditions, the Sci1 T6SS is expressed in only a subset of the population, creating distinct ON and OFF subpopulations in a reversible, heritable, and epigenetically controlled equilibrium. This heterogeneity is governed by the interplay between the iron-responsive regulator Fur- and Dam-dependent DNA methylation at the sci1 promoter. Mutations in Fur binding sites or GATC methylation motifs shift the population to homogeneous ON or OFF states, respectively. Functional analyses reveal that while ON cells mediate antibacterial activity, OFF cells buffer the population against lethal retaliatory responses from defensive T6SS⁺ competitors. Our results suggest that T6SS heterogeneity in EAEC represents a finely tuned attenuation strategy optimizing the trade-off between competitive killing and survival in hostile microbial communities. This work uncovers a novel layer of regulation in T6SS deployment and highlights phenotypic heterogeneity as an adaptive trait in interbacterial warfare.
|
|
Scooped by
mhryu@live.com
June 4, 11:10 PM
|
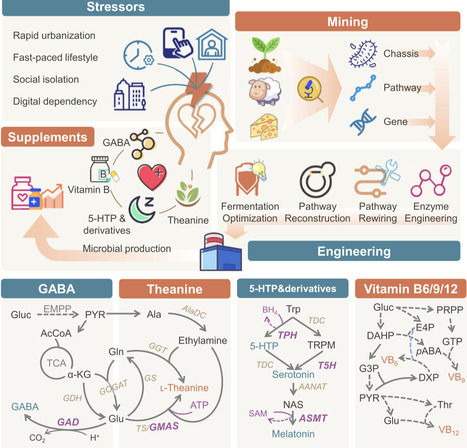
The fast-paced modern life exerts great pressure to individuals. Nutritional interventions for stress management have gained increasing attention due to its favorable safety profiles, multiple health benefits, and suitability for vulnerable populations. Microbial production of stress-relieving nutraceuticals represents a sustainable alternative to natural extraction and chemical synthesis, while meeting the growing market demand for natural-labeled and consumer-preferred products. This review provides an overview of the recent progress in engineering microorganisms to produce common stress-relieving biomolecules, such as: γ-aminobutyric acid, ʟ-theanine, 5-hydroxytryptophan and its derivatives, and vitamin B. Key bottlenecks limiting the bioproduction of each molecules and targeted strategies, including chassis selection, enzyme engineering, pathway rewiring and bioprocess regulation, are discussed.
|
|
Scooped by
mhryu@live.com
June 4, 11:02 PM
|
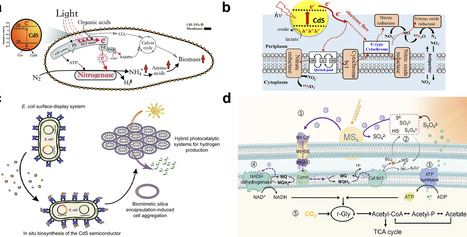
Photosynthetic biohybrid systems (PBSs) integrate semiconductor light harvesters with microbial metabolism to enable solar-driven chemical synthesis, yet the chemical principles governing their performance remain dispersed across two distinct architectures: wired biohybrids, which rely on photoelectrode–microbe interfaces, and wireless systems, where microbes are photosensitized by colloidal or molecular catalysts. This review examines the materials chemistry, interfacial electron transfer mechanisms, and biological constraints that define each approach. We evaluate the stability and biocompatibility of semiconductor photoelectrodes, charge transfer pathways across abiotic/biotic interfaces, microbial community dynamics, and photoelectrochemical operational parameters central to wired systems. For wireless platforms, we analyze design rules for whole-cell photosensitization, including semiconductor selection, cellular uptake, redox coupling, and mechanistic probes of electron delivery. By comparing both architectures, we identify unifying chemical principles and key bottlenecks that limit efficiency, providing a framework for the predictive design of next-generation PBSs for sustainable solar-to-chemical conversion.
|
|
Scooped by
mhryu@live.com
June 4, 10:49 PM
|
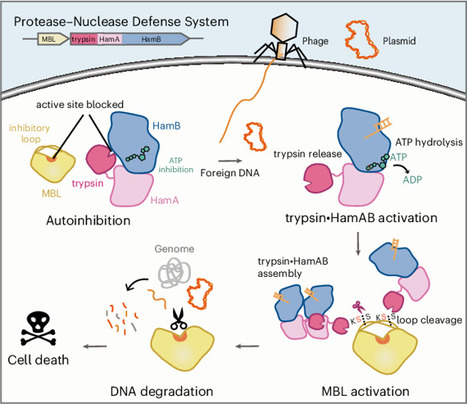
Protease-mediated activation of immune effectors is an evolutionarily conserved mechanism. This study identifies a widespread trypsin–MBL (metallo-β-lactamase) module as a core effector in diverse antiviral bacterial immune systems, such as Hachiman, AVAST and Argonaute. Focusing on the Hachiman-associated trypsin–MBL system, we show that trypsin•HamAB protease activity is inhibited by ATP, while MBL is an autoinhibited DNase with two insertion loops obstructing its catalytic site. Upon infection, trypsin•HamAB senses foreign DNA and hydrolyzes ATP, activating trypsin-like activity, which specifically cleaves MBL at the insertion loops to release repression. The activated MBL depletes DNA and arrests host cell growth. Cryo-electron microscopy structures of trypsin•HamAB–DNA reveal that DNA binding and ATP hydrolysis trigger HamAB oligomerization and trypsin-like domain release, enabling its activation. Our work elucidates a conserved immune mechanism wherein proteolytic activation of a nuclease enables robust immunity against phage while multilayered controls prevent self-toxicity, expanding the repertoire of immune processes governed by regulatory proteolysis. This study identifies and characterizes a conserved trypsin–MBL (metallo-β-lactamase) pair in bacterial immunity; upon infection, trypsin is activated through inhibitory ATP hydrolysis, subsequently activating MBL through site-specific proteolysis, which depletes DNA and restricts cell growth.
|
|
Scooped by
mhryu@live.com
June 4, 5:00 PM
|
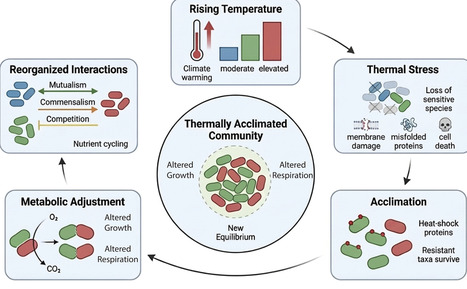
Temperature is a fundamental regulator of microbial physiology, shaping processes from growth and respiration to community interactions and ecosystem functioning. This review synthesizes recent experimental and theoretical advances that reveal how microbes respond to warming across biological scales, with a focus on short-term ecological acclimations of microbial communities. At the cellular level, rising temperature affects enzyme kinetics, membrane fluidity, and metabolic efficiency, often in non-linear ways that challenge the validity of fixed Q10-based models. At the community level, warming tends to favor thermotolerant and slow-growing taxa, while reconfiguring microbial interaction networks by shifting balances between competition, cooperation, and syntrophy. These structural changes can reduce functional redundancy and stability, yet prolonged warming may also foster the emergence of cohesive, resilient community architectures. Overall, we emphasize the need for integrative mechanistic frameworks that link thermal physiology, carbon-use efficiency, and microbial interactions to improve predictions of microbial contributions to carbon cycling under climate change.
|
|
Scooped by
mhryu@live.com
June 4, 4:53 PM
|
Prime editing is a precise and rapid genome-editing technique that modifies short DNA sequences using tailored guide RNAs. To implement this technique in bacteria, we used Prime Editor 2 (PE2) with the DeepPrime gRNA design tool and assessed its gene-editing efficiency in E. coli and methicillin-resistant Staphylococcus aureus (MRSA) cells. Our findings indicate that a split PE2, comprising a reverse transcriptase and two Cas9 nickase domains, exhibited gene-editing efficiency comparable to that of the intact PE2. The efficiency observed in E. coli was significantly affected by the target sites, edit type, and the presence of exonucleases. In MRSA, which serves as a model to evaluate the applicability in non-model bacterial species, Streptococcus pyogenes PE2 (SpPE2) exhibited superior performance relative to Staphylococcus aureus PE2 (SaPE2). Furthermore, the split SpPE2 lacking the reverse transcriptase successfully induced the intended mutation in MRSA. This study demonstrates the feasibility of prime editing within bacterial systems.
|
|
Scooped by
mhryu@live.com
June 4, 4:47 PM
|
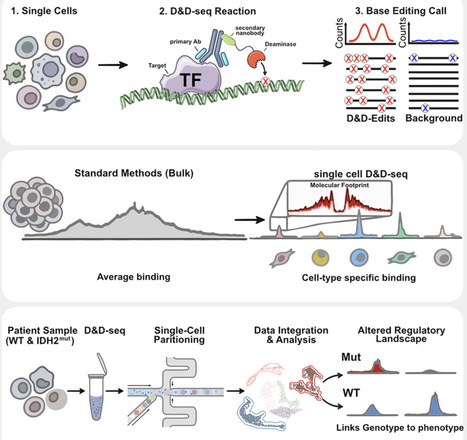
Gene expression is controlled by transcription factors (TFs), whose genome binding is shaped by chromatin accessibility and histone modifications, yet mapping these interactions, particularly those with weak affinity or a transient nature, in single cells remains technically challenging. To address this gap, we developed docking and deamination followed by sequencing (D&D-seq), a single-cell immuno-tethering technology for profiling DNA-protein interactions. D&D-seq couples an antibody-binding nanobody to a cytosine base editor, a combination that enables detection of weak or transient factor binding through targeted cytosine-to-uracil editing at protein-bound genomic sites. This approach is compatible with standard single-cell multi-omic workflows and therefore allows integrated analyses of gene regulation. Using assay for transposase-accessible chromatin using sequencing (ATAC-seq) and single-cell ATAC-seq (scATAC-seq), we assessed chromatin accessibility as a functional readout of TF activity, and by coupling D&D-seq with whole-genome sequencing, we captured CTCF binding in both active and inactive chromatin compartments.
|
|
Scooped by
mhryu@live.com
June 4, 4:33 PM
|
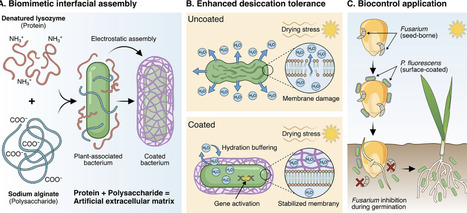
Microbial inoculants are central to sustainable agriculture; however, the vulnerability of bacterial cells to desiccation represents a fundamental barrier to their effective use in open-environment applications. While nature employs extracellular polymeric substances for protection, synthetic replication of this multifunctional, nanoscale interface remains a challenge. Here, we report a biomimetic strategy to assemble an artificial extracellular matrix (AEM) directly on the surface of Pseudomonas fluorescens, conferring exceptional abiotic resilience. Inspired by amyloid-protein architecture in natural biofilms, we engineered an interfacial coating via the conformational transition of lysozyme into a β-sheet-rich, adhesive scaffold, which electrostatically co-assembles with alginate polysaccharides at the cell envelope. This conformal nanocoating provides dual-mode protection: it acts as a viscoelastic hydration buffer that prevents membrane rupture, and it elicits a transcriptional response that upregulates genes associated with respiration, osmoprotection, and proteostasis. Optimized at a 1:1 protein-to-polysaccharide ratio, the AEM enhances bacterial survival after desiccation by 30.9-fold. Furthermore, it enables robust seed adhesion and storage stability, translating into effective biocontrol against Fusarium pathogens in a model agricultural system. This work establishes a versatile strategy for programming cellular interfaces, bridging materials design and microbial functionality to engineer resilient living systems for real-world deployment.
|
|
Scooped by
mhryu@live.com
June 4, 3:10 PM
|
Protein-based biosensors offer unique advantages over conventional analytical methods by enabling real-time detection of target analytes with minimal sample preparation. However, efficiently coupling molecular recognition to a reliable output signal remains a key challenge in biosensor design. Here, we present a plug-and-play strategy using the de novo switch platform, LOCKR, which enables direct transduction of a binding event into a defined signal output. The LOCKR architecture supports modular reconfiguration: the recognition domain can be swapped to detect a desired analyte, while the reporter module can be interchanged to tune the output format. We expand the range of LOCKR-compatible readouts beyond split luciferase to include ratiometric Förster-type resonance energy transfer and β-lactamase-based colorimetry. By integrating computationally designed high-affinity binders as interchangeable recognition elements, we demonstrate sensitive detection of glucagon, neuropeptide Y, and peptide YY with limits of detection in the picomolar range. Together with the expanding landscape of de novo designed binders and reporters, the LOCKR platform bridges the gap between molecular recognition and signal generation, enabling versatile biosensor development tailored for user-defined applications.
|
|
Scooped by
mhryu@live.com
June 4, 1:21 PM
|
Extracellular vesicles are cell-derived secretions that mediate tissue homeostasis and intercellular communication through their diverse cargos, including proteins. Distinct extracellular vesicle biogenesis pathways suggest specific association and co-enrichment of proteins sharing a biogenesis pathway, and non-association and co-depletion of proteins segregated into distinct pathways. Yet these associations elude conventional protein expression or co-expression measurements. Here, we propose and define pairwise protein co-enrichment relative to its overall expression to quantify whether a given protein is co-enriched or co-depleted with another protein. We measure co-enrichment, and differential co-enrichment between a stimulus and a reference condition, of up to 240 protein pairs in extracellular vesicles using antibody microarrays. We validate co-enrichment by modulating well-known extracellular vesicle biogenesis pathways, and find that differential co-enrichment measures expected changes between perturbed and reference conditions. Co-enrichment and differential co-enrichment in three model cell lines and parental and organotropic breast cancer progeny cell lines reveal both preserved and variable co-enrichment that may warrant further studies. Collectively, our result suggest that co-enrichment reflects cell physiology and extracellular biogenesis, is readily measurable, and could serve as quality control in extracellular vesicle biomanufacturing. Distinct extracellular vesicle biogenesis pathways suggest specific co-enrichment of proteins sharing a biogenesis pathway. Here, the authors design an approach to examine this co-enrichment and show that it reflects vesicle biogenesis.
|
|
|
Scooped by
mhryu@live.com
Today, 12:17 AM
|
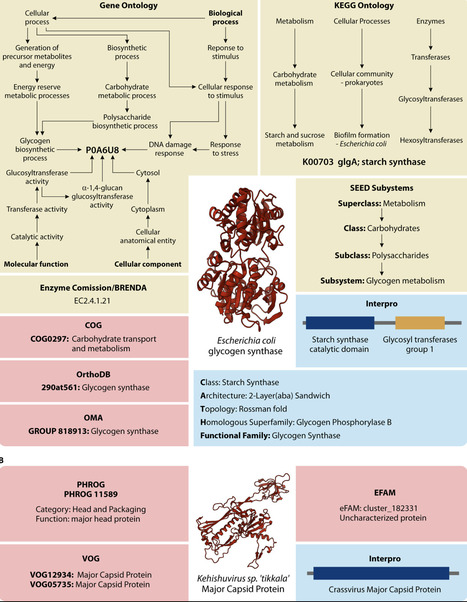
Understanding protein functions is crucial for interpreting microbial life; however, reliable function annotation remains a major challenge in computational biology. Despite significant advances in bioinformatics methods, ~30% of all bacterial and ~65% of all bacteriophage (phage) protein sequences cannot be confidently annotated. In this review, we examine state-of-the-art bioinformatics tools and methodologies for annotating bacterial and phage proteins, particularly those of unknown or poorly characterized function. We describe the process of identifying protein-coding regions and the systems to classify protein functionalities. Additionally, we explore a range of protein annotation methods, from traditional homology-based methods to cutting-edge machine learning models. In doing so, we provide a toolbox for confidently annotating previously unknown bacterial and phage proteins, advancing the discovery of novel functions and our understanding of microbial systems.
|
|
Scooped by
mhryu@live.com
June 4, 11:58 PM
|
Harnessing the innate growth, self-repair, and adaptive capabilities of living systems within engineered devices could transform static buildings via domestic infrastructures into dynamic, self-sustaining platforms. Electroactive biofilms (EABs) provide a unique interface for this vision, naturally converting organic matter into electricity, treating wastewater, and processing complex information. Recent breakthroughs in synthetic biology and artificial intelligence now allow EABs to be programmed as biologically intelligent components—such as living transistors and logic processors—rather than simple biocatalysts. This opinion article outlines a roadmap for transitioning EAB-enabled hybrid biological-artificial systems from laboratory prototypes into integrated architectures for decentralised resource recovery. Ultimately, these bio-intelligent technologies enable a circular economy in which buildings function as metabolic organisms, redefining our relationship with the built environment.
|
|
Scooped by
mhryu@live.com
June 4, 11:38 PM
|
Biosynthetic needs of a cell and the generation of precursors and energy in catabolic reactions must be faithfully balanced. Even in model organisms such as Bacillus subtilis, there are still important gaps in our knowledge. The key bottleneck in research is the lack of novel research hypotheses. New concepts and methodologies can help to develop such hypotheses. Here, we discuss how the introduction of proteome-wide protein-protein interaction mapping by in vivo cross-linking, the AI-mediated prediction of structure models for each protein, and the possibility to compare those models highly efficiently aid the development of novel hypotheses. Moreover, the focused use of suppressor screens can help to get new unbiased insights. We demonstrate how these approaches are applied to B. subtilis. Global cross-linking combined with the power of AI provided a testable hypothesis to unravel the long-standing open question of how iron is sensed in B. subtilis and related bacteria. This is of particular importance as iron is the growth-limiting factor for most bacterial pathogens. The isolation of suppressor mutants that are resistant to growth-inhibiting amino acids has identified novel amino acid exporters. Importantly, the corresponding genes belong to the most poorly expressed genes in B. subtilis, and they are only activated under selective pressure by mutations that affect corresponding transcription factors or the promoter regions of the exporter genes. As the approaches discussed here have only recently been brought to wide application, we can expect that they will be very fruitful in gaining a better understanding of metabolism and metabolic homeostasis.
|
|
Scooped by
mhryu@live.com
June 4, 11:20 PM
|
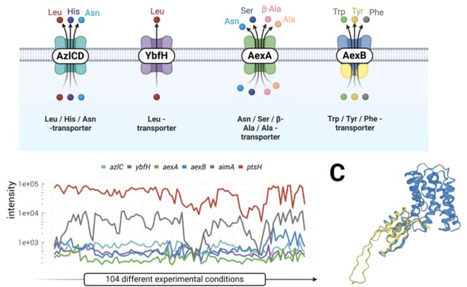
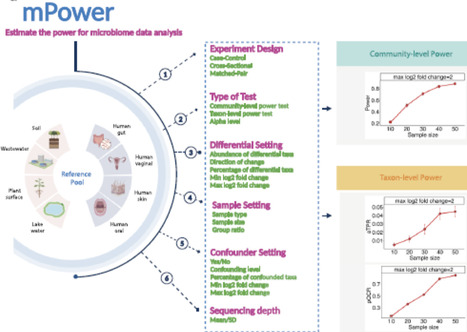
Power analysis is a critical step in designing a microbiome study. Existing power calculation tools for microbiome studies mainly rely on parametric models of the sequencing counts, which underestimate the complexity of microbiome data and could produce overly optimistic power estimates. In this work, we present a new simulation-based power analysis tool, mPower, for microbiome study design. The tool uses a real data-based semi-parametric simulation framework to generate realistic microbiome data, upon which the power assessment is performed. Coupled with a select differential analysis tool, our power tool supports different study designs, including cross-sectional, case-control, and matched-pair studies, with or without confounders. It allows power analysis for both community-level and taxon-level testing. By using microbiome reference datasets from different environments, the users could perform power calculation based on the environment of interest. The mPower is primarily designed for 16S amplicon sequencing data, and it also incorporates a parametric simulation framework that enables power analysis for shotgun metagenomic data. We showcase the application of mPower with several real-world examples. The web interface of mPower is available at https://microbiomestat.shinyapps.io/mPower/. Video Abstract
|
|
Scooped by
mhryu@live.com
June 4, 11:07 PM
|
Natural products (NPs) remain vital to drug discovery, yet the identification of novel bioactive NPs is frequently hampered by the rediscovery of known compounds in traditional screening and the “bioactivity gap” in genome mining. Self-resistance-guided genome mining has emerged as a transformative strategy to address these challenges, leveraging co-localized resistance genes within biosynthetic gene clusters (BGCs) to predict NPs’ molecular targets. This review summarizes recent progress in discovering novel NPs that target essential cellular processes, including protein synthesis, protein degradation, DNA integrity, and primary metabolism. We further highlight key technologies and strategies designed to accelerate this discovery workflow and discuss the limitations and opportunities of self-resistance-guided genome mining for the systematic discovery of precision therapeutics in the genomic era.
|
|
Scooped by
mhryu@live.com
June 4, 10:52 PM
|
Phage therapy often fails when bacteria evolve resistance. We argue that phage selection should begin with receptors whose modification imposes predictable costs, turning resistance into reduced virulence, antibiotic resensitization, or other exploitable trade-offs. Receptor-constrained evolutionary traps offer a framework for designing phages that steer—not merely suppress—bacterial evolution effectively.
|
|
Scooped by
mhryu@live.com
June 4, 5:02 PM
|
In recent years, synthetic biology has been widely applied to engineer and program cellular behaviors. Using this approach, bacteria can be designed to express immunotherapeutic agents, improve tumor targeting, and deliver therapeutic payloads directly to tumor sites. To further improve efficacy, strategies such as hypoxia-responsive promoters, bacterial swarming, and extracellular vesicles (EVs) have been investigated, along with the synergistic effects of combining bacterial therapy with other treatments (e.g., photodynamic therapy, chemotherapy, immune checkpoint inhibitors). This review summarizes recent advances in synthetic biology for bacteria-based cancer immunotherapies, focusing on how bacterial agents activate the immune system and the engineering strategies used to achieve tumor targeting.
|
|
Scooped by
mhryu@live.com
June 4, 4:57 PM
|
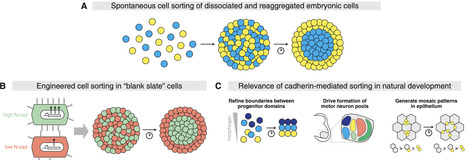
Developmental biology seeks to understand how multicellular organization emerges from cell-cell interactions. Advances in stem cell and synthetic biology now enable researchers to rebuild developmental processes outside the embryo, with varying degrees of resemblance to natural systems. While some reconstituted systems reveal how development occurs, others uncover what is possible. This perspective examines how such bottom-up approaches have elucidated general principles and causal mechanisms of multicellular organization. We argue that synthetic systems, though simplified, provide powerful platforms to test the limits of developmental potential, disentangle causal relationships, and inform predictive models. With rapid advances in genomic engineering, imaging, and computational modeling, leveraging these engineered systems to discover what is possible holds transformative promise for understanding what is happening in nature.
|
|
Scooped by
mhryu@live.com
June 4, 4:51 PM
|
Precise genome editing through targeted DNA insertion is critical for gene therapy and biomedical research. While existing methods rely on homology-directed repair (HDR), this process suffers from low efficiency in non-dividing cells. Microhomology-mediated repair provides an alternative but remains intrinsically inefficient. Here, we developed a DNA polymerase θ (Pol θ)-based editor (PET) to enhance kilobase-scale targeted DNA integration across diverse genomic loci and cell types. We demonstrate that polymerase domain (pPET) and helicase-like domain (hPET) independently improve microhomology-mediated editing, with pPET achieving a 3-fold increase in knock-in rates over conventional SpCas9-mediated editing. Next-generation sequencing reveals that pPET and hPET reduce on-target indel rates by ∼30%, while increasing precise insertions by up to 80%. Compared with other Pol θ domain configurations and MMEJ-enhancing factors, pPET exhibits superior performance. These findings establish Pol θ functional domains as effective tools for improving microhomology-driven genome editing and advancing therapeutic applications.
|
|
Scooped by
mhryu@live.com
June 4, 4:45 PM
|
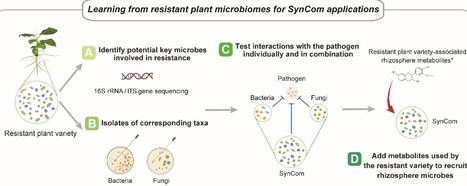
The plant microbiome plays a crucial role in enhancing disease resistance, yet microbiome-based plant protection strategies remain limited by an incomplete understanding of how host selection, microbial interactions, and rhizosphere chemistry jointly shape pathogen suppression. Here, we adopt a “learning from nature” approach to design synthetic microbial communities (SynComs) that recapitulate naturally evolved disease-suppressive interactions, using banana Fusarium wilt as a model system. High-throughput profiling revealed that both bacterial and fungal communities contribute to varietal resistance. Resistance-associated microbial taxa were identified and isolated to assemble bacterial, fungal, and cross-kingdom SynComs representative of resistant versus susceptible hosts. SynComs derived from resistant varieties suppressed pathogen growth more effectively than those from susceptible hosts, with cross-kingdom SynComs exhibiting the strongest effects. Cross-kingdom SynCom inoculation significantly reduced disease severity and restructured both the composition and functional potential of the rhizosphere microbiome. Integrative transcriptomic and metabolomic analyses revealed coordinated host metabolic reprogramming, characterized by increased accumulation of diverse metabolites, including alkaloids, amino acids, and flavonoids. Notably, supplementation with resistance-associated rhizosphere metabolites, such as stearic acid and shikimic acid, further enhanced disease suppression. Together, our findings establish a mechanistic framework in which host-guided microbiome assembly and metabolite-mediated interactions jointly enable effective cross-kingdom SynComs for disease suppression, providing ecological principles for microbiome-based plant protection strategies.
|
|
Scooped by
mhryu@live.com
June 4, 3:25 PM
|
Since its discovery, the CRISPR-Cas system has ushered in a transformative era in biodetection, leveraging its simplicity and efficiency to enable Cas protein-based signaling systems for applications in early tumor screening, viral detection, and molecular logic circuits. However, the constrained compatibility of CRISPR/Cas-based signaling systems with diverse input types limits their versatility, primarily due to the restricted activation mechanisms of the Cas protein. Herein, we developed the Cas12a and Cas13a Integrated Targeting (CACIT) system, which harnesses DNA/RNA strand displacement reactions to integrate the enzymatic capabilities of Cas12a and Cas13a. This system supports simultaneous DNA and RNA inputs, offering exceptional programmability and cost-effectiveness. By employing strand displacement reactions, the CACIT system achieves synchronized activation of Cas12a and Cas13a. We have demonstrated that the CACIT system excels in single-nucleotide-variant (SNV) detection, viral RNA detection, machine learning-driven nucleic acid concentration response modeling, logic operations, and intracellular imaging. As a streamlined and versatile signaling platform, the CACIT system expands the scope of CRISPR/Cas activation strategies. With its inherent simplicity and compatibility, this system facilitates integration with diverse nanodevices. Further, this system provides a highly programmable, multifunctional computational module for molecular networks, heralding new possibilities for artificial signaling systems.
|
|
Scooped by
mhryu@live.com
June 4, 3:06 PM
|
Adenosine triphosphate (ATP) hydrolysis is the main cellular source of energy used to drive biochemical reactions that are otherwise energetically unfavorable. The chemical energy stored in phosphoanhydride bonds is released upon hydrolysis of ATP to ADP and is used to drive mechanical work and conformational change. DNA replication is a canonical process in which the multi-enzyme replisome is thought to rely on ATP hydrolysis for its function. Here we show, through single-molecule visualization of DNA replication by the E. coli replisome, that the replicative DnaB helicase does not rely on hydrolysis of ATP in the context of the elongating replisome. Even in the presence of physiologically-relevant concentrations of ATP, dTTP is hydrolysed preferably. Finally, we show that the replicative helicases from S. cerevisiae, D. melanogaster, and Homo sapiens can also use dTTP to unwind DNA. Our observations suggest that replicative helicases across domains of life are ‘flex-fuel’ helicases. Here the authors show that replicative helicases from bacteria to humans can use dTTP instead of ATP for DNA unwinding, and that the E. coli replisome preferentially uses dTTP during DNA replication, challenging textbook models of replication energetics.
|


















software, microscopy imaging