 Your new post is loading...

|
Scooped by
?
Today, 5:00 PM
|
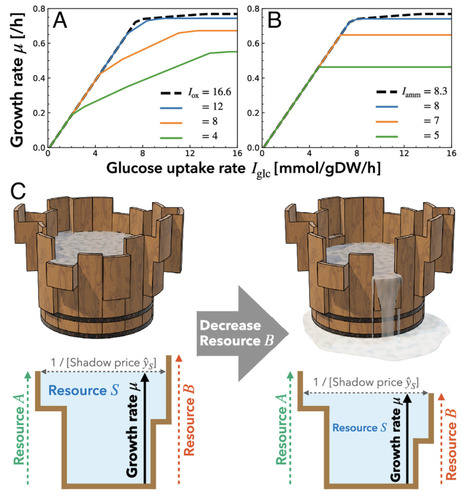
Understanding complex living systems requires identifying universal phenomenological laws that are independent of species and molecular-biological details. The Monod equation is a cornerstone of phenomenological microbial growth laws, depicting the growth rate as a saturating function of a single substrate. Because of its similarity in functional form to the Michaelis–Menten equation, cellular growth is often thought to be limited by a single reaction. However, cellular growth generically results from the coordination of thousands of metabolic reactions and diverse intracellular limited resources. Consequently, the mechanistic origins of the Monod equation remain controversial, and its extension has encountered limitations. Here, we propose the global constraint principle for cellular growth: As one nutrient becomes more available, other intracellular resources become limiting, driving transitions to distinct modes of resource allocation. Based on a general framework of constraint-based modeling and its dual formulations, we mathematically prove that, in general, microbial growth kinetics curves are monotonically increasing and concave with respect to nutrient availability. Numerical simulations using genome-scale Escherichia coli models with proteome allocation, molecular crowding, and membrane capacity constraints reproduce these features in a multiphasic manner. In contrast to the original Monod’s growth law, the global constraint principle also captures the dependence of microbial growth on the availability of multiple nutrients, by generalizing the Liebig’s law of the minimum, another phenomenological growth law for higher organisms, into a terraced landscape of diminishing returns. It thus integrates the classical phenomenological laws proposed by Monod and Liebig into a comprehensive theory of cellular growth.
|
|
Scooped by
?
Today, 4:44 PM
|
Processive endoglucanases, which possess both endo- and exoglucanase activities, are considered highly promising catalysts in cellulose degradation. In this study, we employed multiple deep learning models, including MutCompute, DeepSequence, and ESM-1v, to guide the engineering of EG5C-1, a processive endoglucanase derived from Bacillus subtilis BS-5. This enabled a systematic exploration of the enzyme’s sequence space. Through a combination of clustering analysis and a greedy algorithm, we optimized combinations of amino acid substitutions and ultimately identified an elite variant, M8 (R23Q/E43Q/K91I/K191P/A198T/Q237D/V240P/S245A), composed entirely of substituted residues. Compared to the wild-type enzyme, M8 exhibited 10-fold and 5-fold improvements in catalytic efficiency (kcat/Km) toward soluble substrate carboxymethyl cellulose-Na (CMC) and insoluble substrate phosphoric acid-swollen cellulose (PASC), respectively, along with enhanced optimal temperature and thermostability. Molecular mechanistic analyses revealed that all distal substituted residues enhanced dynamic coupling and coordination, primarily influencing the conformation of three loops near the substrate pocket. These structural changes modulated substrate binding and product release, thereby contributing to improved catalytic efficiency (kcat/Km). This work not only suggests a feasible strategy to explore the “dark space” within sequences but also provides insights into the practical application of machine learning in experiments.
|
|
Scooped by
?
Today, 4:34 PM
|
Coastal bacteria play an important role in the conversion of terrestrial organic carbon (TerrOC). However, their ecological patterns and drivers remains elusive. Here, 180 bacterial communities from 10 regions along the Chinese coastline, covering an 18,000 km transect between 18.27 °N and 39.82 °N, were cultured under three typical lignocellulosic substrates, hardwood (aspen), softwood (pine), and herbaceous (rice straw), respectively. All the consortia showed a broad spectrum of TerrOC utilization, and displayed degradation capacities comparable with those previously established though preliminary in situ lignocellulose enrichment. Moreover, following the metabolic theory of ecology, annual average temperature of the sites stimulated community metabolism, even though all were cultured at 30°C. Consortia enriched on aspen exhibited the highest temperature sensitivity. 16S rRNA gene amplicon and metatranscriptomic sequencing analyses revealed temperature-dependent latitudinal diversity gradients, displaying a trend that was opposite of the temperature-diversity positive relationship observed in terrestrial lignin-degrading microbes. The community composition shifted to adapt to rising environmental temperature. To enhance lignin degradation, aspen consortia from high annual average temperature employed metabolic generalists, which induced expression of dypB centered gene families for lignin depolymerization and versatile pathways for degradation of lignin derivates. This study reveals the intrinsic drivers for coastal cultured lignocellulose degrading bacterial communities from an ecological perspective and deepens our understanding of the metabolic mechanisms in coastal TerrOC conversion.
|
|
Scooped by
?
Today, 4:23 PM
|
The fungal genus Trichoderma has demonstrated significant potential for agricultural applications, particularly as a plant growth promoter and biocontrol agent against various phytopathogens. This study evaluated Trichoderma yunnanense TM10 as a biocontrol agent against Fusarium oxysporum f. sp. cepae (Foc) in shallots through both in vitro and in vivo assays. In vitro assays demonstrated that T. yunnanense TM10 effectively inhibited the growth of Foc through several mechanisms, including mycoparasitism, antibiosis, and competition for space between TM10 and Foc. Gas chromatography-mass spectrometry analysis identified eight volatile organic compounds exhibiting antimicrobial potential, while liquid chromatography-mass spectrometry analysis revealed eighteen non-volatile organic compounds with antifungal properties against Foc. In vivo assays showed that TM10 reduced disease severity by approximately 53.5% and improved plant physiological characteristics, as shown by increased chlorophyll content, carotenoids, phenolic content, and antioxidant enzyme activity. Morphological improvements were also evident, with greater root and leaf elongation, higher biomass accumulation, and a higher number of leaves compared to pathogen-infected controls. These findings suggest that T. yunnanense TM10 controls Foc through multiple mechanisms while promoting plant health, making it a promising and sustainable biocontrol agent for managing Fusarium wilt in shallots.
|
|
Scooped by
?
Today, 11:24 AM
|
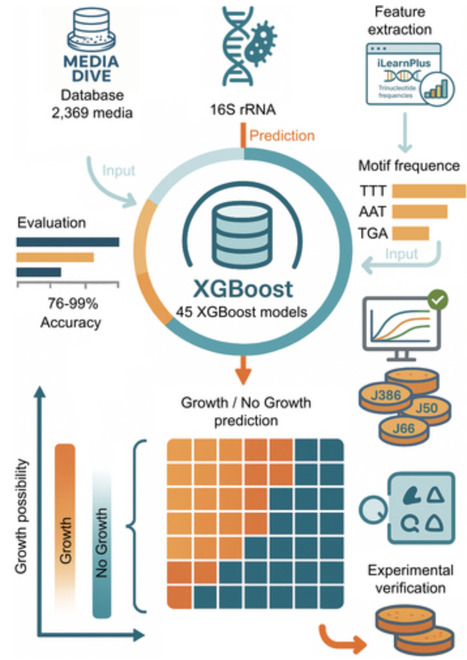
Microorganism culturing is essential in microbiological research, with the selection of suitable culture media being critical for successful microbial growth. Traditionally, this selection has relied on empirical knowledge or trial and error, often resulting in inefficiency. In this study, we analysed nutrient compositions from the MediaDive database to construct a dataset of 2369 media types. Leveraging this dataset and microbial 16S rRNA sequences, we developed 45 binary classification models using the XGBoost algorithm. These models demonstrated strong predictive performance, achieving accuracies ranging from 76% to 99.3%, with the top-performing models for J386, J50 and J66 media reaching 99.3%, 98.9% and 98.8%, respectively. The models effectively predicted growth conditions for various human gut microbes, confirming their practical utility. This research improves the efficiency of microbial cultivation and highlights the potential of machine learning to optimise culture media selection and advance microbiological studies.
|
|
Scooped by
?
Today, 11:13 AM
|
The genomic signatures of microbial domestication remain poorly understood within the context of natural population variation. Here, we demonstrate that Aspergillus oryzae, the filamentous fungus used in soy sauce production, shares more recent ancestry with a predominantly northern, largely non-aflatoxigenic population of Aspergillus flavus (population C). Strikingly, A. oryzae isolates also overlap with the recently described, clinically enriched A. flavus population D, suggesting the possibility of multiple domestication events. Although A. oryzae exhibits reduced virulence compared to A. flavus, all isolates tested retained pathogenicity in a zebrafish infection model. At the transcriptomic level, Aspergillus populations are significantly differentiated, with distinct responses to population density, indicating that population-specific transcriptomes adapt differently to ecological conditions. These differences extend beyond gene content and are not always explained by phylogenetic relationships, suggesting that phenotypic diversification occurs through the rapid reorganization of transcriptomic architectures. For example, A. oryzae displays significantly elevated expression of a module enriched for carbohydrate metabolism. Population-specific variation is also evident among secondary metabolite (SM) gene clusters. While A. oryzae shows markedly reduced expression of specific SM genes, particularly those involved in aflatoxin biosynthesis, this trend does not extend across the entire secondary metabolome. Using machine-learning-based gene regulatory network inference, we identified population-specific transcriptomic differences linked to distinct transcription factors, with evidence for both cis- and trans-acting regulatory divergence, but no changes in global regulators such as laeA. Together, these findings provide new insights into the domestication of A. oryzae, its global significance, and the microevolution of fungal secondary metabolic pathways.
|
|
Scooped by
?
Today, 10:29 AM
|
As the ability to synthesize complete genomes becomes increasingly accessible, the question of what should compose those sequences is becoming more prevalent. However, identifying genes essential for the survival of an organism is challenging, as gene essentiality is a nuanced concept that heavily depends on context. In this study, we identified growth medium-specific important genes by performing transposon mutagenesis in Escherichia coli BW25113 and sequencing mutant populations at multiple time points in three growth media. Our analysis revealed a core set of 412 important genes shared across all conditions, along with distinct medium-specific gene sets of varying sizes. By analyzing temporal variations in read counts for each gene, we identified additional sets of genes whose inactivation causes a lighter impact on fitness. We used this dataset to define medium-specific gene modules required to sustain robust growth under each condition. Our study underscores the context-dependent nature of gene essentiality and marks a step toward refining the concept from a universal list to a more nuanced, condition-specific framework, which will be invaluable for future genome design efforts.
|
|
Scooped by
?
October 3, 7:34 PM
|
Plasmids are a foundational research reagent and a key material in biopharmaceutical manufacturing. Plasmids require a backbone for propagation in E. coli, which typically contains an antibiotic resistance gene alongside a replication origin. As plasmids increasingly enter clinical applications, concerns are raised on the safety risks of antibiotic resistance genes. Additionally, these protein-coding genes occupy long stretches of DNA that incur significant metabolic burdens on host cells, which negatively impacts plasmid manufacturability and functionality, leading to high production cost and compromised clinical efficacy. Here, we describe miniVec, a novel miniaturized plasmid backbone devoid of protein-coding sequence, and instead expresses a small RNA to provide constant selective pressure capable of sustaining high plasmid copy numbers in plain culture media devoid of antibiotics or other chemical additives. This simplifies large-scale fermentation and greatly increases plasmid yield. Notably, miniVec confers enhanced functionality in a variety of applications such as chemical transfection, electroporation, virus packaging, transposon- or CRISPR-mediated genome integration, and in vivo naked DNA transfection and vaccination, while exhibiting no detectable immunogenicity or toxicity. These advantages establish miniVec as the next-generation plasmid platform for clinical applications, featuring improved safety that aligns with regulatory expectations, enhanced manufacturability leading to much higher yield and dramatic cost reduction, and augmented functionality in diverse applications.
|
|
Scooped by
?
October 3, 7:22 PM
|
The identification of complex spatial patterns of microbial communities in relation to their ecological niches is fundamental to understanding the mechanisms of ecological interactions among diverse organisms. This study introduces a novel three-dimensional (3D) sampling approach to examine the spatial dynamics of microbial populations and niche differentiation influenced by plant-mediated effects in natural ecosystem. Microbial communities across horizontal and vertical dimensions were systematically mapped, and we found that the total microbial diversity, particularly among eukaryotes, increased more than ten-fold compared to that obtained via single-grid sampling, emphasizing the importance of spatial heterogeneity in shaping microbial dynamics. Moreover, the 3D framework enabled us to identify taxa specifically associated with particular plants, offering insights into plant–microbe interactions, pathogen prevalence, and ecological consequences of plant-driven effects on local communities. Collectively, these findings demonstrate that 3D sampling approach provides a reproducible and scalable methodology for investigating microbial spatial heterogeneity, pathogen ecology, and niche differentiation in natural environments.
|
|
Scooped by
?
October 3, 7:12 PM
|
Flavonoid O-glycosylation, catalyzed by uridine diphosphate (UDP)–glycosyltransferases, is crucial for their therapeutic efficacy. However, most UDP-glycosyltransferases encounter three major limitations: low activity, poor regioselectivity, and restricted substrate availability, hindering their pharmaceutical applications. To address these challenges, we conducted protein engineering on a previously unidentified glycosyltransferase, UGT75AJ2, which had 3′,7-O-glycosylation capabilities. Our approach involved three strategies: (i) development of a tailored focused rational iterative site-specific mutagenesis strategy, augmented by virtual screening and iterative mutagenesis, to design mutant Mut4-1 (S367A/V274A/F82V/I132T) with a 128-fold enhancement in relative catalytic activity; (ii) enhancement of the enzyme’s compatibility with a broader spectrum of sugar donors achieved through structural-based engineering, yielding mutant S14G/F366H/S367G and demonstrating effective utilization of diverse donors; (iii) construction of a targeted mutant library to enhance regioselectivity by active site analysis, leading to mutants with high selectivity for targeted glycosylation sites. This comprehensive study tackles predominant challenges in UDP-glycosyltransferases protein engineering, providing innovative approaches and insights that enhance the development of flavonoid glycosides.
|
|
Scooped by
?
October 3, 6:54 PM
|
Targeted degradation is emerging as a new therapeutic approach in the treatment of different diseases. It allows hijacking the cellular pathways deputed to protein or nucleic acid homeostasis to degrade a target macromolecule of interest involved in a pathogenic process. In the last decades, targeted protein degradation has been widely applied for the treatment of cancer or neurodegenerative disorders and some of such therapies are already in clinical use. More recently, therapeutic degraders such as PROTACs, LYTACs, HyTs, BacPROTACs, and others have also been explored in the field of antimicrobial and antiviral drug discovery. The peculiar mechanism of action, along with the opportunity to degrade both microbial and host targets, holds great promise for overcoming some limitations of classic antimicrobials, e.g. drug resistance, as well as for increasing the potency of current therapies. With a focus on the antimicrobial field, this Review aims at providing a comprehensive, state-of-the-art description of targeted degradation mechanisms and strategies developed so far, as well as to discuss advantages, disadvantages, and caveats of this innovative approach for combating infectious diseases.
|
|
Scooped by
?
October 3, 1:23 PM
|
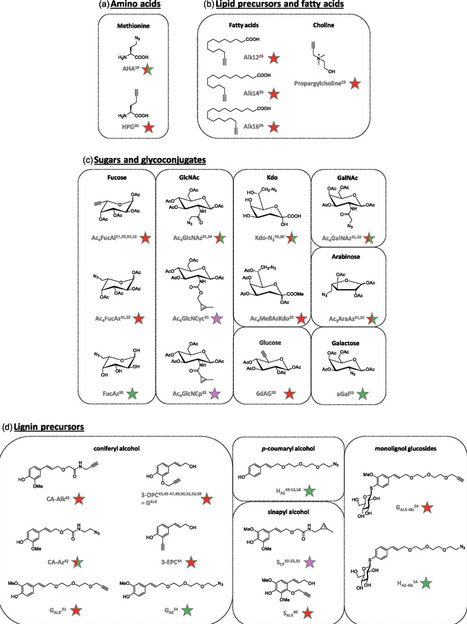
Chemical biology has reshaped the ability to investigate complex biological systems at the molecular level. In this context, chemical reporters have become important tools for labeling and tracking biomolecules in living systems with spatial and temporal precision. In plant biology, they provide an alternative to genetic approaches and allow the study of dynamic processes in species or organs that are not easily accessible. Through the use of click and bioorthogonal chemistry, small-molecule probes can be metabolically incorporated into specific molecular scaffolds such as sugars, monolignols, amino acids, and lipids. These probes make it possible to follow events like glycosylation, lignification, lipid turnover, or protein synthesis in living plant tissues. This review presents an overview of current chemical reporter strategies, from molecular design and synthetic considerations to their application in plant imaging. Herein, how these tools have contributed to the development of plant chemical biology by enabling precise and modular investigations of plant structure and metabolism is described. Herein, it is also examined how chemical reporters have entered interdisciplinary contexts, including collaborations between science and the arts. By converting molecular-level information into visual and sensory formats, these approaches open new perspectives for research, education, and communication across scientific and creative disciplines.
|
|
Scooped by
?
October 3, 10:53 AM
|
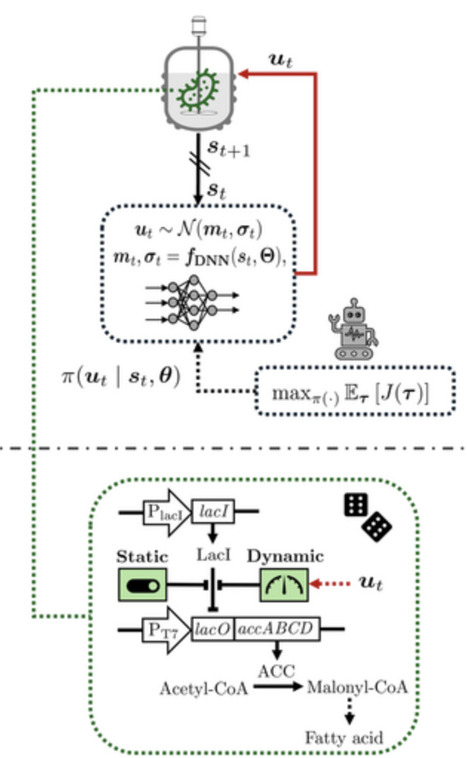
Dynamic metabolic control allows key metabolic fluxes to be modulated in real time, enhancing bioprocess flexibility and expanding available optimization degrees of freedom. This is achieved, for example, via targeted modulation of metabolic enzyme expression. However, identifying optimal dynamic control policies is challenging due to the generally high-dimensional solution space and the need to manage metabolic burden and cytotoxic effects arising from inducible enzyme expression. The task is further complicated by stochastic dynamics, which reduce bioprocess reproducibility. We propose a reinforcement learning framework to derive optimal policies by allowing an agent (the controller) to interact with a surrogate dynamic model. To promote robustness, we apply domain randomization, enabling the controller to generalize across uncertainties. When transferred to an experimental system, the agent can in principle continue fine-tuning the policy. Our framework provides an alternative to conventional model-based control such as model predictive control, which requires model differentiation with respect to decision variables; often impractical for complex stochastic, nonlinear, stiff, and piecewise-defined dynamics. In contrast, our approach relies on forward integration of the model, thereby simplifying the task. We demonstrate the framework in two Escherichia coli bioprocesses: dynamic control of acetyl-CoA carboxylase for fatty-acid synthesis and of adenosine triphosphatase for lactate synthesis.
|
|
|
Scooped by
?
Today, 4:47 PM
|
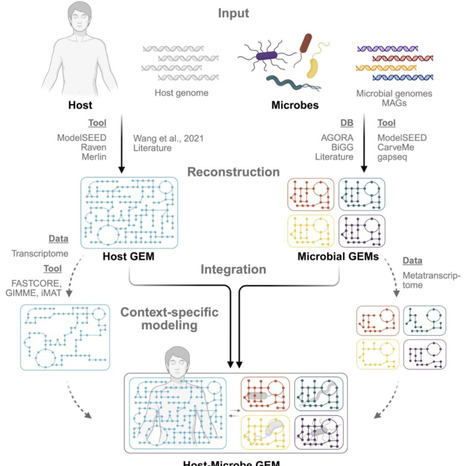
Host-microbe interactions play an integral role in the function and survival of eukaryotes, influencing various processes ranging from metabolism to immune regulation. As our understanding of these interactions deepens, there is a growing shift toward integrative approaches that consider both host and microbial genotypic potential. However, capturing the complexity and dynamic nature of these relationships remains a significant challenge. Genome-scale metabolic models (GEMs) offer a powerful framework to investigate host-microbe interactions at a systems level. By simulating metabolic fluxes and cross-feeding relationships, GEMs enable the exploration of metabolic interdependencies and emergent community functions. These models can be applied independently or in conjunction with experimental data, supporting hypothesis generation and systems-level insights into host-microbe dynamics. In this review, we examine recent applications of GEMs to host-microbe studies, with a focus on how they reveal reciprocal metabolic influences. We also discuss the current technical challenges, highlight available tools and methodological strategies, such as model reconstruction, data integration, and simulation and analysis steps, and outline future directions for advancing host-microbe interaction study using GEMs.
|
|
Scooped by
?
Today, 4:39 PM
|
Cellular desiccation—the loss of nearly all water from the cell—is a recurring stress that drives widespread protein dysfunction. To survive, part of the proteome must resume function upon rehydration. Which proteins tolerate desiccation, and the molecular determinants that underlie this tolerance, are largely unknown. Here, we use quantitative mass spectrometry and structural proteomics to show that certain proteins possess an innate capacity to tolerate extreme water loss. Structural analyses point to protein surface chemistry as a key determinant of desiccation tolerance, which we test by showing that rational surface mutants can convert a desiccation-sensitive protein into a tolerant one. We also find that highly tolerant proteins are responsible for the production of small-molecule building blocks, while intolerant proteins are involved in energy-consuming processes such as ribosome biogenesis. We propose that this functional bias enables cells to kickstart their metabolism and promote cell survival following desiccation and rehydration.
|
|
Scooped by
?
Today, 4:30 PM
|
The development of novel antimicrobial agents that are effective against Gram-negative bacteria is hindered by the dual membrane cell envelope of these bacteria. To reach their intracellular targets, most small-molecule antibiotics must first pass through protein channels called porins; however, a common mechanism of acquired resistance is decreased expression of these outer membrane proteins. Additionally, parameters such as size, shape, and charge regulate passage of antibiotics through porins, further limiting the design space of novel antibiotic molecules. Inspired by the ability of bacterial outer membrane vesicles (OMV) to deliver cargo to the bacterial cytosol, we hypothesized that encapsulation of small molecule antibiotics within OMVs would improve the activity of the drugs by facilitating uptake. To test this hypothesis, we investigated the ability of imipenem-encapsulated OMVs to inhibit the growth of several Gram-negative bacteria, including multidrug-resistant (MDR) clinical isolates. Our results demonstrated that encapsulation within OMVs significantly lowers the effective concentration of imipenem in several MDR isolates. Using a panel of porin knockout strains, we further demonstrated that this mechanism of antibiotic delivery does not require porin expression. Together, our results demonstrate the potential of OMVs as novel antibiotic delivery vehicles to treat antibiotic-resistant bacterial infections by improving drug uptake.
|
|
Scooped by
?
Today, 4:13 PM
|
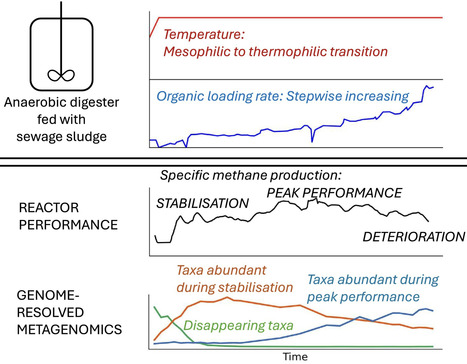
This study investigated temporal dynamics in reactor performance and microbial community structure during anaerobic digestion of sewage sludge when the temperature was changed from 37°C to 55°C, followed by an increase in organic loading rate (OLR). Performance instability was observed immediately following the temperature increase and in the end of the study when the OLR was 11.1 ± 0.3 kgVS m−3d−1. The specific methane production peaked at 0.31 ± 0.06 Nm3 kg−1 volatile solids (VS) during thermophilic operation and when the OLR was 3.5 ± 0.9 kgVS m−3d−1. Using metagenomic sequencing, 304 species-representative genome bins (SGB) were assembled. Network analysis revealed that 186 SGB were associated with thermophilic conditions and several new species putatively involved in key reactor functions were identified. When reactor function initially stabilised, two hydrogenotrophic and one aceticlastic methanogen (Methanothermobacter spp. and Methanosarcina thermophila), the hydrolytic Coprothermobacter proteolyticus, and putative syntrophic propionate oxidisers (e.g., Pelotomaculaceae) had high relative abundance. During the peak in specific gas production, the community was dominated by one hydrogenotrophic Methanothermobacter species coexisting with syntrophic acetate oxidizing bacteria (Thermacetogenium phaeum and other species). Finally, when the reaction function deteriorated due to high OLR, new hydrolytic taxa emerged and the same aceticlastic methanogen as seen during the initial acclimatisation phase returned.
|
|
Scooped by
?
Today, 11:19 AM
|
Lysine acylations are a diverse class of post-translational modifications (PTMs) that dynamically regulate protein function. Access to homogenous, site-specifically modified proteins is essential for dissecting their molecular roles, yet remains challenging with traditional methods. Here we present a modular strategy that combines genetic code expansion (GCE) with a chemoselective amide bond-forming reaction to install a wide range of lysine acylations directly onto folded proteins. Key to this approach is the genetic encoding of Nϵ-methoxylysine, which reacts efficiently with N-methyliminodiacetyl (MIDA) acylboronates to generate diverse acyl lysine modifications from a single protein precursor, including several acyl PTMs not previously accessible through GCE. This mild and broadly applicable methodology enables systematic studies of lysine acylation on multiple target proteins. We demonstrate its utility by probing the effects of defined acyl modifications on enzymatic activity, protein-RNA interactions and deacylase-mediated regulation. Together, this platform establishes a versatile strategy to interrogate the functional consequences of this wide-spread class of PTMs.
|
|
Scooped by
?
Today, 10:40 AM
|
The structural integrity of bacterial cells is traditionally attributed to the peptidoglycan cell wall, and more recently to the outer membrane, with the cytoplasmic membrane assumed to be mechanically passive. Cells lacking filaments of the actin homolog MreB are more bendable, suggesting a role for the cytoskeleton in cell stiffness. Here, we show that MreB does not stiffen the envelope directly, but instead mechanically couples the cell wall to the cytoplasmic membrane through its role in peptidoglycan synthesis, increasing resistance to bending. Under hyperosmotic stress, MreB relocalized to the poles, forming linkages that prevent membrane detachment from the cell wall and attenuate cytoplasmic contraction. Disruption of MreB filament formation, nutrient starvation, or inactivation of glycan elongation factors abolished or reduced this coupling, revealing that peptidoglycan biosynthesis actively mediates stress distribution across surface layers. Our findings redefine the bacterial envelope as a mechanically integrated composite, with the cytoplasmic membrane having substantial load-bearing capacity.
|
|
Scooped by
?
October 3, 7:58 PM
|
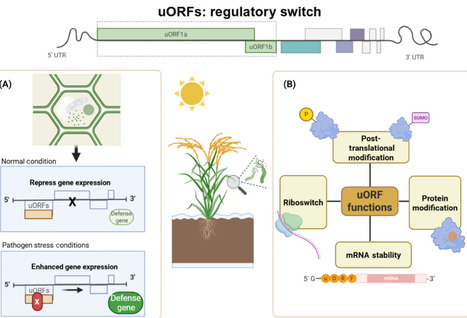
The discovery and manipulation of upstream open reading frames (uORFs) represent a promising frontier in plant biotechnology, offering strategies to enhance disease resistance and crop resilience. Here, we explore the role of uORFs in regulating gene expression under biotic stress and discuss approaches to engineer uORFs for sustainable agriculture and precision breeding.
|
|
Scooped by
?
October 3, 7:29 PM
|
The discovery of novel antimicrobial peptides (AMPs) against clinical superbugs is urgently needed to address the ongoing antibiotic resistance crisis. AMPs are promising candidates due to their broad-spectrum activity, rapid bactericidal mechanisms and reduced likelihood of inducing resistance compared with conventional antibiotics. Here, a pre-trained protein large language model (LLM), ProteoGPT, was established and further developed into multiple specialized subLLMs to assemble a sequential pipeline. This pipeline enables rapid screening across hundreds of millions of peptide sequences, ensuring potent antimicrobial activity and minimizing cytotoxic risks. Through transfer learning, we endowed the LLMs with different domain-specific knowledge to achieve high-throughput mining and generation of AMPs within a unified methodological framework. Notably, both mined and generated AMPs exhibited reduced susceptibility to resistance development in ICU-derived carbapenem-resistant Acinetobacter baumannii (CRAB) and methicillin-resistant Staphylococcus aureus (MRSA) in vitro. The AMPs also showed comparable or superior therapeutic efficacy in in vivo thigh infection mouse models compared with clinical antibiotics, without causing organ damage and disrupting gut microbiota. The mechanisms of action of these AMPs involve disruption of the cytoplasmic membrane and membrane depolarization. Overall, this study presents a generative artificial intelligence approach for the discovery of novel antimicrobials against multidrug-resistant bacteria, enabling efficient and extensive exploration of AMP space. This study presents a generative artificial intelligence approach for the high-throughput discovery of antimicrobials against multidrug-resistant bacteria.
|
|
Scooped by
?
October 3, 7:20 PM
|
Chromosome architecture plays a crucial role in bacterial adaptation, yet its direct impact remains unclear. Different bacterial species and even strains within the same species exhibit diverse chromosomal configurations, including a single circular or linear chromosome, two circular chromosomes, or a circular-linear combination. To investigate how these architectures shape bacterial behavior, we generated near-isogenic strains representing each configuration in Agrobacterium tumefaciens C58, an important soil bacterium widely used for plant genetic transformation. Strains with a single-chromosome architecture, whether linear or circular, exhibited faster growth, enhanced stress tolerance, and greater inter-strain competitiveness. In contrast, bipartite chromosome strains showed higher virulence gene expression and enhanced transient plant transformation efficiency, suggesting a pathogenic adaptation. Whole-transcriptome analysis revealed architecture-dependent gene expression patterns, underscoring the profound impact of chromosome organization on Agrobacterium fitness and virulence. These findings highlight how chromosome structure influences bacterial adaptation and shapes evolutionary trajectories.
|
|
Scooped by
?
October 3, 7:00 PM
|
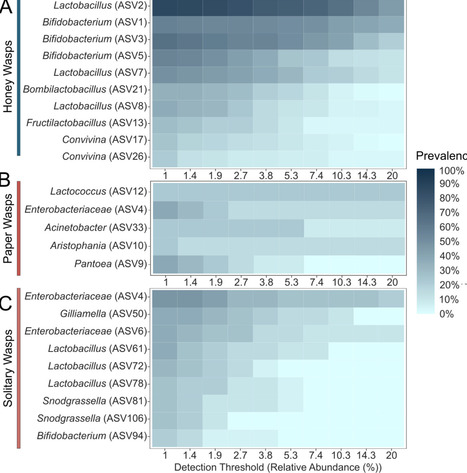
Honey-feeding social bees, including honey bees, bumble bees, and stingless bees, possess distinctive gut bacterial communities that provide benefits to hosts, such as defense against pathogens and parasites. Members of these communities are transmitted through social interactions within colonies. The Mexican honey wasp (Brachygastra mellifica) represents an independent origin of honey-storing within a group of social Hymenoptera. Honey wasps feed on and store honey, but, unlike bees, they prey on other insects as a protein source and do not consume pollen. We surveyed the gut bacterial communities of Mexican honey wasps across sites within Texas using 16S ribosomal RNA profiling, and we estimated bacterial titer per bee using quantitative PCR. For comparison, we also surveyed non-honey-feeding wasps from six families, collected in the same region. We found that honey wasp communities are dominated by characteristic bacterial species. In contrast, other wasps had lower absolute titers and more variable communities, dominated by environmental bacteria. Honey wasps from all sampled nests contained strains of Bifidobacterium and Bombilactobacillus that were closely related to symbionts of bumble bees and other bees, suggesting acquisition via host-switching. Some individuals also harbored a close relative of Candidatus Schmidhempelia bombi (Orbaceae), an uncultured bumble bee symbiont, again suggesting host-switching. The most prevalent species was an uncultured Lactobacillus that potentially represents an independent acquisition of environmental Lactobacillus. The transition to honey feeding, combined with a highly social life history, appears to have facilitated the establishment of a bacterial community with similarities to those of social bees.
|
|
Scooped by
?
October 3, 5:11 PM
|
Uncovering genotype-phenotype relationships is hampered by genetic redundancy. For example, most genes in Streptococcus pneumoniae are non-essential under laboratory conditions. A powerful approach to unravel genetic redundancy is by identifying gene-gene interactions. We developed a broadly applicable dual CRISPRi-seq method and analysis pipeline to probe genetic interactions (GIs) genome-wide. A library of 869 dual single-guide RNAs (sgRNAs) targeting high-confidence operons was created, covering over 70% of the genetic elements in the pneumococcal genome. Testing these 378,015 unique combinations, 4,026 significant GIs were identified. Besides known GIs, we found previously unknown positive and negative interactions involving genes in fundamental cellular processes such as division and chromosome segregation. The presented methods and bioinformatic approaches can serve as a roadmap for genome-wide gene interaction studies in other organisms. All interactions are available for exploration via the Pneumococcal Genetic Interaction Network (PneumoGIN), which can serve as a starting point for new biological discoveries.
|
|
Scooped by
?
October 3, 12:32 PM
|
Reactive oxygen species (ROS), inevitable byproducts of aerobic respiration, exhibit both signaling and cytotoxic effects depending on concentration within cells. At controlled levels, ROS serve as crucial signaling molecules mediating intracellular signal transduction pathways. However, excessive accumulation triggers oxidative stress, leading to oxidative damage that disrupts protein structure and function, ultimately inducing cell death. Enhancing cellular adaptive responses to oxidative stress and mitigating intracellular ROS accumulation are therefore critical strategies for maintaining cellular homeostasis. This approach significantly improves product yield and underpins its widespread application across diverse sectors, including functional foods, pharmaceuticals, and cosmeceuticals. This review first delineates the origins and multifaceted roles of ROS. Subsequently, it details strategies employed to bolster cellular oxidative stress resilience, focusing on process optimization and metabolic engineering approaches. Finally, synthesizing current research advances, existing challenges, and emerging trends, the review outlines future research directions aimed at enhancing the oxidative stress tolerance of microbial cell factories.
|


























2st