 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 9:29 AM
|
lthough protein engineering and laboratory evolution have been used to optimize prime editors, we show that previous changes that improve prime editor efficiency also compromise protein stability and expression level, limiting performance. To address these limitations, we apply structure-informed artificial intelligence-guided methods such as the inverse-folding network ProteinMPNN to redesign the reverse transcriptase (RT)domains of engineered and evolved prime editors while preserving regions essential for catalysis. Redesigned RTs are extensively mutated, with 30–163 amino acid substitutions, and exhibit enhanced folding stability and soluble expression and up to twofold higher intracellular prime editor protein levels following mRNA delivery. Redesigned PE8 prime editors demonstrate enhanced editing efficiencies across multiple ex vivo contexts, including in several human primary cell types and via several delivery modalities. In mice, editing efficiency is up to 2.9-fold higher than that of state-of-the-art PE6, PE7 and PEmax prime editors. These findings demonstrate a generalizable approach for augmenting laboratory evolution to improve genome editing agents. A computational redesign strategy improves evolved prime editors.
|
|
Scooped by
mhryu@live.com
Today, 12:13 AM
|
Quantitative analysis of bacterial dynamics in time-lapse microscopy requires robust tracking pipelines, yet selecting and optimizing algorithms for specific experiments remains challenging. Indeed, microbiologists are confronted with numerous algorithms that must be carefully chosen and parameterized to achieve optimal tracking for their experiments. We present an automated methodology to determine optimal tracking configurations for microbiological applications. It is based on TrackMate 8, a novel version of the TrackMate Fiji plugin extended with microbiology-specific tools. Our approach systematically evaluates algorithm-parameter combinations optimizing biologically relevant metrics (e.g., cell-cycle accuracy, bacterial morphology) and includes: (1) integration of deep-learning algorithms (Omnipose, YOLO, Trackastra) adequate for bacterial images in TrackMate; (2) a TrackMate-Helper extension for parameter optimization; and (3) a tracking and segmentation editor for tracking ground-truth generation. We demonstrate the effectiveness of the methodology on two use cases showing its adaptability to diverse experimental conditions. This methodology enables microbiologists with a widely applicable, automated framework to optimize tracking pipelines, facilitating quantitative analysis in bacterial imaging.
|
|
Scooped by
mhryu@live.com
Today, 12:01 AM
|
Plants actively reshape the soil environment through their roots and associated microbes, creating lasting changes known as soil legacies that influence future plant generations via plant–soil feedbacks. While biotic factors such as pathogens and mutualists have received much attention, the chemical legacies, including water-soluble and volatile organic compounds, remain underexplored. These metabolites, produced by plants and soil microbes, modulate microbial communities, nutrient dynamics, and plant defenses, driving positive or negative feedbacks. This opinion article synthesizes recent evidence on soil chemical diversity, their role in legacy formation and persistence, while highlighting analytical challenges and promising applications in agriculture and ecology.
|
|
Scooped by
mhryu@live.com
May 20, 5:13 PM
|
The artificial intelligence (AI)-driven generation of genetic sequences holds transformative potential for addressing global challenges in agriculture, medicine, and bioenergy. Traditional approaches including hybridization, mutagenesis, and CRISPR-based editing enable targeted modification of endogenous DNA, yet remain constrained by natural sequence diversity. We here introduce PlantGFM, an application of the Hyena operator within a plant-oriented genomic foundation model, which was pre-trained on 10.84 billion nucleotides from 12 plant species and supports long-context (64 kb) prediction and sequence generation within a unified architecture. After fine-tuning on 10 annotated plant genomes, PlantGFM matched or exceeded the performance of specialized gene prediction tools. Beyond reproducing natural genes, it enables de novo design of novel candidates through the emergence capability of AI. Seven candidates selected through an AI–Human Knowledge fusion screening pipeline all showed transcriptional activity in Nicotiana benthamiana, two with stable protein expression—representing the first demonstration of DNA–RNA–protein expression of Large Language Model-generated sequences in plants. As a proof of concept, PlantGFM also exhibits emergent abilities in generating plant NLR genes. Our findings establish the feasibility of LLM technology for de novo plant gene design, providing a foundation for plant synthetic biology and AI-assisted breeding.
|
|
Scooped by
mhryu@live.com
May 20, 5:08 PM
|
Immobilised Metal Affinity Chromatography (IMAC) is widely used to purify his-tagged recombinant proteins from E. coli. However, endogenous contaminants with histidine clusters, such as GFAT and PDH E1 proteins, are often co-purified with the target protein. The low background strain LOBSTR-RIL has been previously engineered with mutated forms of SlyD and ArnA that exhibit reduced binding to Ni2+ resin. In this study, the LOBSTR-RIL strain was further modified to produce IMACulate(DE3), where we altered the glmS (encoding GFAT protein) and aceE (encoding PDH E1 protein) genes to reduce surface histidines. Proteins purified from this strain show reduced levels of GFAT contamination. No statistically significant difference was observed in the abundance levels of PDH E1 protein in the BL21(DE3)-RIL, LOBSTR-RIL and IMACulate(DE3) strains. The use of IMACulate(DE3) increases the purity of recombinant his-tagged protein preparations with no additional effort or expense.
|
|
Scooped by
mhryu@live.com
May 20, 5:02 PM
|
Pseudomonas aeruginosa and Klebsiella pneumoniae are gram-negative opportunistic pathogens that frequently colonize the human body and are major causes of infection. These bacteria are often co-isolated in polymicrobial urinary tract and lung infections, the latter of which is associated with increased disease severity and worse clinical outcomes. Despite their overlapping niches and clinical relevance, little is known about how these two pathogens interact and how those interactions influence human health. Given the growing recognition that microbial interactions are key drivers of disease, we investigated how P. aeruginosa and K. pneumoniae influence one another. We discovered an antagonistic interaction in which P. aeruginosa restricts the growth of K. pneumoniae. This inhibition is driven by phenazine production in P. aeruginosa, specifically the secondary metabolites pyocyanin and pyorubin, which are both necessary and sufficient to suppress K. pneumoniae growth. Using a diverse set of clinical isolates, we found that this antagonism is strain-dependent. Both the susceptibility of K. pneumoniae to phenazines and the ability of P. aeruginosa to restrict K. pneumoniae growth varies between strains. Moreover, the necessity of phenazine production is specific to the site of infection. Together, these findings demonstrate that strain background and environmental context are critical determinants of pathogen interactions. These findings reveal that both strain background and environmental redox conditions govern the ecological rules of pathogen interaction, providing a framework for predicting outcomes.
|
|
Scooped by
mhryu@live.com
May 20, 4:53 PM
|
Drug-resistant bacterial infections increasingly evade available antimicrobials, and many existing antimicrobial peptides remain limited by instability, toxicity to mammalian membranes and high manufacturing cost. Here we introduce a modular peptide technology that self-assembles into nanofibres on bacterial surfaces through a membrane-anchoring biphenyl group, a diphenylalanine linker and a cationic minimalistic peptide that together enable selective disruption of drug-resistant pathogens. Using cryogenic electron microscopy, molecular dynamics simulations, lipid-nanoparticle membrane mimetics and binding thermodynamics, we show that the peptide first forms short nanofibres that dock onto phosphatidylglycerol and then elongates into nanofibres that penetrate and destabilize the bacterial membrane without inducing resistance. The nanofibres retain antibacterial activity when recycled from killed bacteria and outperform vancomycin and several classical antimicrobial peptides against dense bacterial populations in vitro. In a mouse model of methicillin-resistant Staphylococcus aureus pneumonia, inhaled peptide nanofibres eradicate pulmonary infection and restore lung architecture without detectable toxicity. This modular strategy enables the design of potent, selective and low-cost antimicrobials. A modular peptide technology that self-assembles into nanofibres on bacterial surfaces through a membrane-anchoring biphenyl group, a diphenylalanine linker and a cationic peptide enables disruption of drug-resistant pathogens.
|
|
Scooped by
mhryu@live.com
May 20, 4:42 PM
|
Plants and animals respond to pathogens through pattern recognition receptor and Nod-like receptor proteins. Pathogens commonly use protein effectors to suppress host immunity for successful infection. However, the existence of non-protein effector classes remains comparatively understudied. Here we report an RNA–RNA recognition mechanism governing pathogen–host interaction, mediated by a regulatory RNA-encoding DNA sequence that separately generates two complementary regulatory RNAs. Specifically, a long non-coding RNA transcribed from this DNA region in the fungal pathogen Magnaporthe oryzae translocates into host rice cells and sequesters a complementary microRNA (miRNA), derived from a distinct host DNA region, thereby subverting host immunity. In turn, this rice-derived miRNA promotes disease resistance by repressing the expression of PKR1, a gene that encodes a negative regulator of host immunity. Sequestration of the host miRNA by the fungal long non-coding RNA releases PKR1 expression to facilitate fungal infection. We discovered that this regulatory RNA-encoding DNA sequence is probably widely present across diverse life species, mediating interactions between pathogens and their plant hosts. Collectively, our findings provide an approach for effective disease control using miRNAs derived from this important DNA region. A fungal long non-coding RNA from Magnaporthe oryzae translocates into rice cells to sequester a host microRNA that normally represses PKR1, a negative immunity regulator, thereby facilitating infection and revealing a widespread RNA-based pathogen–host interaction mechanism.
|
|
Scooped by
mhryu@live.com
May 20, 4:27 PM
|
Temperate bacteriophages are ubiquitous viruses that co-evolve with their bacterial hosts. They are defined by their ability to undergo two distinct life cycles: the lytic cycle, in which the phage produces more viral copies and kills the host, and the lysogenic cycle, in which the temperate phage exists as a prophage in the host. Temperate phages have long served as fundamental models in microbiology, genetics and evolutionary biology research, and their life cycles are among the most thoroughly characterized in virology. Historically, the phage life cycle was viewed primarily through the lens of how excision, replication and packaging drive the formation of infective particles. Although it captures the central processes of the phage life cycle, this narrow perspective overlooks the full range of interactions with the host and with other mobile genetic elements. In this Review, we re-examine the temperate phage life cycle in light of emerging insights that expand on this framework with unanticipated complexities. We argue that many properties of (pro)phages should be viewed as integral parts of their life cycle instead of being discussed separately. This holistic view is important to fully appreciate the intricacies of the temperate phage life cycle and the key roles of these viruses in microbiology and biotechnological applications. The life cycle of temperate bacteriophages involves lytic or lysogenic cycles and has historically served as a model for studying genetic regulation. This Review provides an updated overview of these cycles and highlights their complexities, supporting a greater appreciation of the ecological roles of temperate phages.
|
|
Scooped by
mhryu@live.com
May 20, 4:14 PM
|
Staphylococcus aureus is a leading cause of lethal bacteremia and pneumonia, which are driven by potent virulence factors such as T-cell superantigens and alpha hemolysin. S. aureus has among the highest rates of antibiotic resistance, yet no vaccines or alternative therapies are available. Here, we developed a repertoire of potent, high-affinity nanobodies (Nbs) targeting key toxins in S. aureus infection, including Hla and superantigens SEB, SEC, and TSST-1. Comprehensive cryo-EM and AlphaFold3 analyses of these Nbs, which were elicited with clinical cocktail vaccines, revealed diverse neutralizing epitopes and mechanisms that provide insights for immunotherapy and vaccine strategies. Guided by these findings, we engineered stable, multivalent, and multifunctional Nb constructs. These constructs included an aerosolizable trimeric Nb with enhanced neutralization activity against Hla and SEC, and a decameric Nb-IgG-Fc fusion construct with pM or better potencies against a wide range of major toxins in S. aureus sepsis (SEB, SEC, TSST-1, and Hla). These multifunctional Nbs demonstrated protective activity in murine models of pneumonia and sepsis, underscoring their potential as versatile immunotherapies that address the complex virulence of S. aureus. Our work lays a foundation for precision immunotherapies beyond current treatment options to combat complex bacterial infections with multiple virulence mechanisms. Here, the authors develop a repertoire of multivalent nanobodies that target key toxins in Staphylococcus aureus infection that show protective activity in murine models of sepsis and pneumonia.
|
|
Scooped by
mhryu@live.com
May 20, 3:29 PM
|
The evolution of the eukaryotic cell paved the way for the emergence of all complex life on Earth. Despite its significance, the environmental context of early eukaryote evolution is largely unknown. Here we use the geological record to reconstruct the habitats of the oldest known fossil eukaryotes, approximately 1.75–1.4 billion years old. Our integrated palaeontological, sedimentological and geochemical analyses show that although fossil eukaryotes are found in samples deposited in a range of environments from coastal to offshore, they are almost entirely restricted to those from settings with oxygenated bottom waters. This distribution suggests these organisms were aerobes (obligate, facultative and/or microaerophilic) and, given their size and morphological complexity, probably possessed mitochondria. Furthermore, their near absence from otherwise fossiliferous anoxic samples suggests a benthic habit, as planktonic eukaryotes would be expected to be present in both oxic and anoxic samples. We propose that eukaryotes were largely restricted to oxic benthic habitats for much of the Proterozoic eon, only expanding into planktonic habitats during the Neoproterozoic era (1–0.54 billion years ago). This late ecological expansion could account for the mismatch between the appearance of eukaryotic body fossils and molecular biomarkers and explain the stepwise increase in eukaryote diversity during the Neoproterozoic era. Integrated palaeontological, sedimentological and geochemical analyses of ancient rocks from Australia show that early eukaryotes were largely restricted to oxygenated benthic habitats, probably possessed mitochondria by 1.75 billion years ago, and may have expanded into planktonic habitats much later, perhaps during the Neoproterozoic era.
|
|
Scooped by
mhryu@live.com
May 20, 2:56 PM
|
Obesity is associated with metabolic disorders due to unhealthy white adipose tissue (WAT) failing to sustain energy homeostasis, highlighting the importance of adipose development. The lactation period is critical for epididymal WAT (eWAT) development and metabolic programming. However, the role of lactoferrin (LF) in the early development of adipose remains unclear. Using a mouse model of lactational LF deficiency and single-nucleus RNA sequencing, we assessed the long-term impact of LF deficiency on eWAT plasticity and metabolic homeostasis at weaning, adulthood, and under a high-fat diet (HFD). LF deficiency persistently impaired eWAT development, causing restricted adipocyte hyperplasia, exacerbated hypertrophy, diminished lipid uptake, and sustained adiponectin decline with resistin elevation. These defects led to long-term metabolic disorders, worsening HFD-induced eWAT remodeling, glucose intolerance, dyslipidemia, and chronic inflammation. Mechanistically, LF could bind CSK and PRMT5. LF promoted CSK degradation, activating SRC to drive preadipocyte proliferation. Additionally, LF stabilized PRMT5 to enhance PPARg-mediated differentiation and lipid uptake. Rescue experiments confirmed that CSK overexpression reversed LF-induced proliferation, while PRMT5 knockdown blocked LF-enhanced differentiation. This study reveals lactational LF as a key nutritional signal that programs adipose development and long-term metabolic health via CSK-SRC and PRMT5-PPARg pathways, offering an early-life intervention strategy against obesity-related metabolic diseases.
|
|
Scooped by
mhryu@live.com
May 20, 12:51 AM
|
Metagenomes offer the potential to characterize E. coli strain-level diversity within the human gut microbiome, informing our understanding of colonization diversity and the genetic features distinguishing infection from carriage. Among numerous reference-based tools for short-read metagenomic strain-level profiling, the best approach remains unclear. Here, we benchmarked six published tools—PanTax, PathoScope, StrainGE, Strainify, StrainR2 and StrainScan—for their ability to detect co-existing strains of E. coli and estimate their relative abundance across real and simulated metagenomes of increasing complexity with varying reference database composition. In the ZymoBIOMICS® D6331 dataset, only PanTax achieved zero error when predicting the equal abundance of five E. coli strains. In a differentially abundant four-strain mock community dataset (SRR13355226), StrainScan had the lowest mean absolute proportional error (0.89), driven by reduced sensitivity (0.5), followed by PathoScope (4.08). Across simulated metagenomes reflecting the healthy adult gut microbiome, all tools demonstrated high sensitivity (≥0.833), but specificity, precision and F1 score were selectively improved in some tools through detection thresholds to remove low abundance false positives. Outright, StrainGE achieved the highest F1 score (0.978). Predicted relative abundances of the E. coli K12-MG1655 (phylogroup A) and O157:H7 Sakai (phylogroup E) strains spiked into simulated metagenomes across varying abundance ratios were generally accurate, with PanTax and StrainR2 showing the lowest mean absolute proportional error (0.06). When truly present strains were removed from the reference database, out-of-phylogroup assignments were observed for some tools. Collectively, our results demonstrate that published metagenomic strain-level profiling tools vary in their ability to profile E. coli strains, indicating that method selection should be guided by intended application. These findings will facilitate characterisation of E. coli strain-level diversity within short-read gut metagenomes with greater accuracy than previously possible.
|
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Language models trained on biological sequences are advancing inference tasks from the scale of single proteins to that of genomic neighborhoods. Here, we introduce ProteomeLM, a transformer-based language model that uniquely operates on entire proteomes from species spanning the tree of life. ProteomeLM is trained to reconstruct masked protein embeddings using the whole proteomic context, yielding contextualized protein representations that reflect proteome-scale functional constraints. Notably, ProteomeLM’s attention coefficients encode protein–protein interactions (PPI), despite being trained without interaction labels. Furthermore, it enables interactome-wide PPI screening that is substantially more accurate, and orders of magnitude faster, than amino acid coevolution-based methods. We further develop ProteomeLM-PPI, a supervised model that combines ProteomeLM embeddings and attention coefficients to achieve state-of-the-art PPI prediction across benchmarks and species. Finally, we introduce ProteomeLM-Ess, a supervised gene essentiality predictor that generalizes across diverse taxa. Our results demonstrate the potential of proteome-scale language models for addressing function and interactions at the organism level.
|
|
Scooped by
mhryu@live.com
Today, 12:08 AM
|
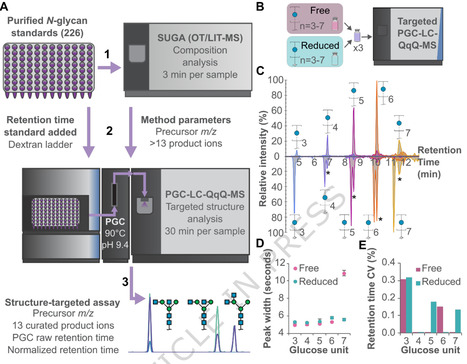
Measurements of glycans modifying glycoproteins are hampered by the lack of standards that reflect the wide diversity in structure typically observed. To this end we exploit a large library of N-glycan standards comprised of a unique collection of 226 N-glycans including oligomannose, hybrid, and complex-type and apply a method employing porous graphitised carbon (PGC) and liquid chromatography mass spectrometry (PGC-LC-MS) to provide a high-resolution separation and characterization of underivatized N-glycan structures. Chromatogram libraries arising from this study include retention time data, diagnostic fragments, and validated structural assignments, providing a robust platform for both targeted and discovery-based glycomics. Here we establish this generated data as an N-glycopedia, the resource in which researchers can compare this collective data to N-glycans under study and overcome the limitations of only having compositional data and predicted structures. The technology is easily expandable to include additional N-glycans as new standards become available. Researchers created N-glycopedia, a reference library of 226 sugar molecules found on proteins, enabling a native glycomics method to precisely identify and measure these sugars, which influence immunity and disease, without chemical pre-treatment.
|
|
Scooped by
mhryu@live.com
May 20, 11:50 PM
|
Plastic waste such as polyethylene terephthalate (PET) is a major environmental burden, and enzymes capable of degrading PET are emerging as biocatalytic tools for sustainable recycling. Progress in improving PET hydrolases (or PETases) has been constrained by the lack of simple and reliable screening systems. Here, we report a functional screen for PETase activity in E. coli based on a zone-clearing assay, where the YebF secretory pathway is used to secrete YebF-PETase onto agar plates supplemented with bis(2‑hydroxyethyl) terephthalate (BHET). Enzyme activity is observed as zones of clearance around E. coli colonies that express active YebF-PETases, as insoluble BHET is converted to soluble products. As proof of concept, the screen was used to evaluated libraries of YebF-LCC-PETase generated by site‑saturation mutagenesis at the active site residues Y95, L102, and V212. This led to the identification of the more active LCC-PETase variants V212T and L102F‑V212T. The secretion‑based assay was then validated using turbidity assays and untagged LCC‑PETase variants, where the L102F‑V212T variant was confirmed to be more active against PET than the wild type enzyme. Docking simulations indicated that V212T improves substrate positioning in the active site while L102F modifies surface charge and hydrophobicity, potentially enhancing binding to the hydrophobic substrate. Overall, the YebF secretion‑driven functional screen serves as a straightforward platform for identifying improved PETase variants and potentially other plastic degrading enzymes.
|
|
Scooped by
mhryu@live.com
May 20, 5:10 PM
|
This investigation revealed that the application of specific microbial inoculants could facilitate the effective biodegradation of polyethylene terephthalate (PET) microplastics collected from urban garbage sites, resulting in non-toxic end products. We employed Azotobacter chroococcum (MTCC 3853), Rhizobium leguminosarum (MTCC 9766), Azospirillum brasilense (MTCC 4036), and Trichoderma viride (MTCC 9681) for PET microplastics degradation and assessed their degradation efficacy through a series of controlled in vitro batch experiments. The study encompassed quantitative analysis of PET weight loss, detailed chemical profiling of degradation intermediates and products, biofilm formation assessment, microbial growth monitoring, and measurement of plastic-degrading enzyme induction. To comprehensively evaluate environmental safety, phytotoxicity assays were performed on Vigna mungo and Vigna radiata, while zebrafish embryos and adults were subjected to acute and embryonic toxicity tests. A. chroococcum (MTCC 3853) was identified as the most efficient strain, showing the greatest reduction in PET mass, enhanced biofilm formation, sustained microbial growth, and peak enzymatic activity, with no detrimental effects on plant or aquatic models, confirming the safety of the biodegradation process. These results underscore the potential of A. chroococcum (MTCC 3853) as a powerful and environmentally friendly solution for microplastic remediation in urban environments.
|
|
Scooped by
mhryu@live.com
May 20, 5:04 PM
|
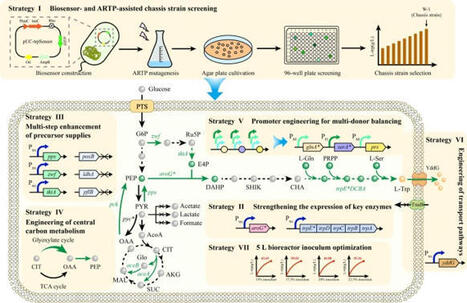
L-Tryptophan is an important aromatic amino acid with wide applications across the food, pharmaceutical, and feed industries. However, its efficient microbial production remains challenging due to complex metabolic networks and multi-level feedback regulation. In this study, we constructed a highly efficient E. coli cell factory for L-tryptophan biosynthesis by combining systematic metabolic engineering with high-throughput screening. Initially, a tnaC-based biosensor was developed and coupled with atmospheric and room temperature plasma (ARTP) mutagenesis to isolate high-performance chassis strains. Central carbon metabolism was subsequently reprogrammed to minimize carbon loss and channel metabolic fluxes toward essential precursors, phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P). To further alleviate pathway bottlenecks, promoter engineering was utilized to balance the intracellular supplies of L-glutamine, L-serine, and phosphoribosyl pyrophosphate (PRPP). This targeted intervention yielded a 21.61% increase in L-tryptophan accumulation. Product transport systems were then engineered to enhance extracellular secretion and mitigate intracellular toxicity. Following the optimization of inoculum size and feeding strategies in a 5 L bioreactor, the final engineered strain (W-24) produced 50.83 g/L of L-tryptophan within 40 h, achieving a yield of 0.185 g/g glucose. This multi-modular engineering framework establishes a robust platform for L-tryptophan biosynthesis and provides a scalable strategy for the industrial production of other valuable aromatic compounds.
|
|
Scooped by
mhryu@live.com
May 20, 4:58 PM
|
Plant pathogens possess about twice as many chemoreceptors as the bacterial average, suggesting broad chemotactic capacities. The signals recognized by most phytopathogen chemoreceptors are unknown, and the reasons for this elevated chemoreceptor number is unclear. We identified the signals recognized by three chemoreceptors, PacH, PacI and PacG, in the global phytopathogen Pectobacterium atrosepticum. The ligand-binding domains (LBDs) of these chemoreceptors share modest sequence similarity, but the signals they recognize are structurally similar, and their biosynthetic pathways are interwoven. Whereas PacH and PacI recognized benzoate derivatives, including salicylate, vanillin and p-hydroxybenzoate, PacG bound agmatine, feruloylagmatine and p-coumaroylagmatine. These compounds are known plant defense compounds, their production is induced by pathogen attack, and they typically accumulate at infection sites. All compounds, except agmatine, induced chemoattraction, which was abolished by mutations in the corresponding genes. Agmatine competed with feruloylagmatine and p-coumaroylagmatine for PacG-LBD binding in vitro and antagonized chemotaxis in vivo. A mutant in pacG, but not in other chemoreceptor genes, showed reduced virulence in planta. We report high-resolution structures of PacG-LBD that were used for ligand-docking experiments to identify its binding pocket. PacH, PacI and PacG homologs were identified in other important phytopathogens belonging to the Burkholderia, Erwinia, Ralstonia, Pectobacterium and Dickeya genera. This is the first report of chemotaxis to feruloylagmatine, p-coumaroylagmatine and p-methoxybenzoate, expanding the range of chemoeffectors. Bacteria thus exploit plant defense responses by moving to compounds that are secreted at infection sites in response to pathogen attack. Chemotaxis to plant defense compounds may be a means to access infected plants and infection sites.
|
|
Scooped by
mhryu@live.com
May 20, 4:43 PM
|
The performance of prime-editing (PE) systems has been improved by systematic engineering of their protein and small RNA components but the structured RNA motifs appended to the 3′ end of PE guide RNAs (pegRNAs)—a key determinant of pegRNA stability and editing efficiency—have not been extensively studied. We introduce PE-PRISM, a high-throughput pooled screen to identify and optimize these 3′ RNA motifs in human cells. Here, using PE-PRISM, we evaluated 2,858 RNA motifs across four iterative libraries, including natural and engineered pseudoknots, G-quadruplexes and reverse transcriptase recruitment elements. We applied structure-guided mutagenesis and combinatorial variant screening to refine hits, culminating in the engineered and evolved pseudoknot variants tevo2.0, eHAV and eSBRMV1-A. In a screen correcting 847 pathogenic ClinVar variants, the top-performing motifs improved PE efficiency over the widely used tevopreQ1 motif for >90% of edits. They also increased PE efficiencies for correcting disease-associated mutations in primary human cells and in vivo in mouse brain and liver. Prime editing is improved with engineered RNA-stabilizing motifs.
|
|
Scooped by
mhryu@live.com
May 20, 4:32 PM
|
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are natural products with diverse structures and functions. Here, we report the discovery of a family of RiPPs whose biosynthetic gene clusters are widespread in the Bacillota genomes and often co-localize with those of lasso peptides, another distinct family of RiPPs. The synthesis of both kinds of RiPPs relies on specific interactions between small adapter protein domains known as RiPP recognition elements (RREs) with their precursor peptides. As these latter share a conserved RRE-binding motif, conflicts between the two biosynthetic pathways may emerge. Through biochemical and structural studies, we reveal how the two RiPP biosynthetic systems evolved to discriminate between their cognate precursors and leader peptidases, allowing them to coexist within a single host. Thus, our study provides insights into the evolutionary diversification of RiPP families. One bacterium can produce many ribosomally synthesized and post-translationally modified peptides (RiPPs) from different families. Here, the authors show how two closely related RiPP families in Bacillota co-evolved to exclude non-cognate precursor peptides, preventing biosynthetic pathway interference.
|
|
Scooped by
mhryu@live.com
May 20, 4:24 PM
|
Genetically encoded fluorescent biosensors (GEFBs) are invaluable tools for spatiotemporal metabolite monitoring in cellular metabolism, yet their development for many key metabolites is hampered by a lack of specific biorecognition elements. Here, we report a versatile strategy to engineer metabolite-responsive GEFBs by leveraging the allosteric properties of regulatory domains from allosteric enzymes. Using regulatory domains from chorismate mutase, 2-acetolactate synthase, and d-citramalate synthase as biorecognition elements, we construct three biosensors for specific l-phenylalanine, l-valine, and l-isoleucine detection. We further demonstrate that multi-ligand-binding regulatory domains can be exploited to derive diverse specific biosensors, and apply this strategy to develop two S-adenosyl-l-methionine biosensors and an S-methyl-5’-thioadenosine biosensor. We also showcase the utility of these biosensors for real-time, in situ tracking of target metabolites in living cells, as well as bioprocess monitoring and clinical diagnostics. Overall, this study establishes a flexible strategy that provides insights to construct GEFBs targeting other metabolites. Genetically encoded fluorescent biosensors are valuable tools for metabolite monitoring. Here, authors report a versatile strategy to design such biosensors via regulatory domains of allosteric enzymes, showing that the developed tools support sensitive detection of target metabolites in vitro and in live cells.
|
|
Scooped by
mhryu@live.com
May 20, 3:58 PM
|
Although the largest completely symmetric closed assembly that can be built from a single building block is the 60-subunit icosahedron, viruses can form capsid assemblies with hundreds to thousands of identical subunits through quasisymmetry—using the same subunit in symmetrically non-equivalent locations in the assembly. Quasisymmetric one-component assemblies could have considerable advantages for delivery of biologics because of the large internal volume achieved using only a single building block, but the design of these structures is challenging because of the inherent complexity of designing chemically identical subunits to both adopt different conformations and make different interactions in the distinct symmetrically non-equivalent locations. Here we conjectured that quasisymmetry could arise from spontaneous symmetry breaking in a system of strongly interacting building blocks with programmed curvatures and show that this principle, coupled with a design approach combining a parametric representation of cage architecture with RoseTTAFold diffusion generative modelling, can generate a rich array of quasisymmetric assemblies. Electron microscopy confirmed the structures of designed 3 ≤ T ≤ 36 cages with 180–2,160 subunits and diameters from 68 nm to 220 nm, and designed 1 < T < 3 non-icosahedral clathrin-like assemblies. Cryogenic electron microscopy structure determination showed how the global symmetry breaking associated with the formation of both hexons and pentons in the T = 3 architecture arises from symmetry breaking in the designed subunit interface. Our results indicate how the detailed architecture of complex systems can be controlled by designing overall system properties, and our approach provides a roadmap for designing large quasisymmetric assemblies for biologics delivery and other applications. Quasisymmetry could arise from spontaneous symmetry breaking in a system of strongly interacting building blocks with programmed curvatures, and this principle, coupled with a design approach, can generate a rich array of quasisymmetric assemblies.
|
|
Scooped by
mhryu@live.com
May 20, 3:03 PM
|
UV exposure causes not only cytosine-to-thymine (C > T) substitutions in dipyrimidine sequences but also many non-canonical mutation classes (e.g. A > T, T > C, and AC > TT). While C > T substitutions are thought to arise from mutagenic bypass of cyclobutane pyrimidine dimers (CPDs), the photoproduct(s) that cause non-canonical mutation classes, which are responsible for key driver mutations in melanoma, are unclear. Here, we use lesion-specific photolyases and whole genome sequencing of yeast irradiated with predominately UVB light to dissect the origins of different classes of UV mutations. These data reveal that CPDs are responsible for ∼60% of all UV mutations and cause not only C > T mutations but also a subset of non-canonical mutation classes, particularly in CT sequence contexts. Our data also indicate that UV-induced pyrimidine (6–4) pyrimidone photoproducts (6–4PPs) are responsible for slightly less than half of all UV-induced mutations in yeast and are the primary cause of T > C substitutions in TT sequences and C > A substitutions. Finally, ∼5% of UV mutations are resistant to both CPD and 6–4PP photolyases, and these are comprised of A > T and AC > TT substitutions in purine-containing dinucleotides. Collectively, these findings define the photoproducts responsible for different UV mutation classes across a eukaryotic genome and indicate that many A > T and AC > TT substitutions arise from atypical UV photoproducts.
|
|
Scooped by
mhryu@live.com
May 20, 2:52 PM
|
Generative neural networks enable de novo design of synthetic genetic elements. Here, we present DiPro, an unconditional diffusion model that learns E. coli promoter features to generate novel sequences without conditional input. DiPro directly generates 50-bp promoter sequences from Gaussian noise using a time-conditioned Transformer denoiser, without auxiliary discriminators or recognizers. DiPro accurately recapitulates natural promoter statistics, including sequence motifs, k-mer spectra, conserved −35/−10 spacing (16–18 bp). Over 90% of generated sequences were classified as real promoters by an independent recognizer, indicating high structural fidelity. To design stationary-phase–specific promoters, generation was guided by inpainting randomly masked regions (three non-overlapping 6-mers, 18 bp) within sigma38-associated (σ38) seed sequences, yielding 5000 candidates while preserving global promoter architecture. Bayesian classification resolved these sequences into three distinct clusters with divergent −35/−10 composition and motif conservation. Experimentally, 81.6% of tested promoters exhibited stationary-phase activity, with the strongest variant reaching 79.6% of the activity of the constitutive promoter J23119. Promoters displayed distinct activation timing, reflecting heterogeneous stress responses. As a proof-of-concept, selected promoters were used to drive a lysis protein, achieving phase-specific and tunable cell lysis that matched promoter activation timing, with early-phase promoters reducing culture density to an optical density at 600 nm of approximately 1.5–2.0. Together, these results establish DiPro as a generative framework for creating functional bacterial promoters with programmable temporal dynamics, offering a new tool for synthetic biology and metabolic engineering.
|































1str