Your new post is loading...

|
Scooped by
?
Today, 2:59 PM
|
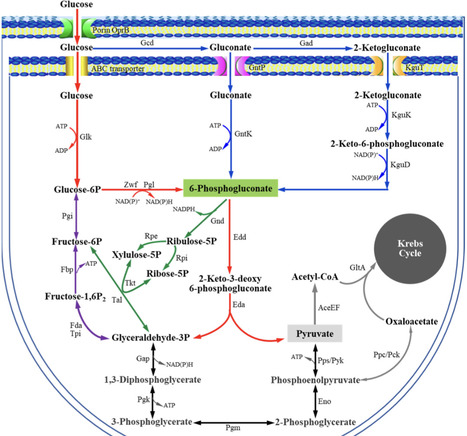
The GntR is a transcriptional regulator generally known as a gluconate-operon repressor to specifically regulate the transportation and phosphorylation of gluconate. In the present study we report the cloning of the GntR-encoding gene of the industrial 2-ketogluconate (2KGA)-producer Pseudomonas plecoglossicida JUIM01, which is involved in the regulation of gluconate metabolism, along with the identification of some of its target genes and its operator sequence. GntR is a 36.36-kDa cytoplasmic and hydrophobic DNA-binding transcriptional regulator belonging to the LacI family. The knockout of gntR resulted in the significant upregulation of the transcription of the gluconate kinase gene gntK and, to a lesser extent, the permease gene gntP, as well as downregulation of genes involved in glucose uptake (oprB-1, gltB, gltF, gltG, and gltK) and those involved in 2-ketogluconate (2KGA) transport (kguT) and catabolism (kguE, kguK, and kguD). These results indicated that GntR positively regulated glucose and 2KGA transport and catabolism, while negatively affecting GntP-mediated gluconate uptake and gluconate phosphorylation by GntK. Electrophoretic mobility shift assay (EMSA) and DNase I footprinting analyses confirmed that GntR interacted with operator sequences in the divergent promoter regions of gntK and gntP, as well as in the gntR promoter region. A putative operator sequence (consensus 5′-AG-N2-AGCGCT-N-TCT-3′) was identified. These data suggest that GntR positively regulates genes involved in glucose uptake/transport and 2KGA transport/catabolism, while repressing its own expression as well as that of genes involved in gluconate transport/catabolism. These findings not only elucidate the regulation of GntR and its target genes in P. plecoglossicida, but also provide valuable insights for optimizing industrial 2KGA production.
|
|
Scooped by
?
Today, 2:51 PM
|
Advancing protein design is crucial for breakthroughs in medicine and biotechnology. Traditional approaches for protein sequence representation often rely solely on the 20 canonical amino acids, limiting the representation of non-canonical amino acids and residues that undergo post-translational modifications. This work explores discrete diffusion models for generating novel protein sequences using the all-atom chemical representation SELFIES. By encoding the atomic composition of each amino acid in the protein, this approach expands the design possibilities beyond standard sequence representations. Using a modified ByteNet architecture within the discrete diffusion D3PM framework, we evaluate the impact of this all-atom representation on protein quality, diversity, and novelty, compared to conventional amino acid-based models. To this end, we develop a comprehensive assessment pipeline to determine whether generated SELFIES sequences translate into valid proteins containing both canonical and non-canonical amino acids. Additionally, we examine the influence of two noise schedules within the diffusion process–uniform (random replacement of tokens) and absorbing (progressive masking)–on generation performance. While models trained on the all-atom representation struggle to consistently generate fully valid proteins, the successfully generated proteins show improved novelty and diversity compared to their amino acid-based model counterparts. Furthermore, the all-atom representation achieves structural foldability results comparable to those of amino acid-based models. Lastly, our results highlight the absorbing noise schedule as the most effective for both representations. https://github.com/Intelligent-molecular-systems/All-Atom-Protein-Sequence-Generation
|
|
Scooped by
?
Today, 2:33 PM
|
Characterizing virus–host pairs and the infection state of individual cells is the major technical challenge in microbial ecology. We addressed these challenges using state-of-the-art single-cell genome technology (SAG-gel) combined with extensive metagenomic datasets targeting the bacterial and viral communities in Lake Biwa. From two water layers and two seasons, we obtained 862 single-cell amplified genomes (SAGs), including 176 viral (double-stranded DNA phage) contigs, which identified novel virus–host pairs involving dominant freshwater lineages. The viral infection rate, estimated by mapping the individual SAG’s raw reads to viral contigs, showed little variation among samples (12.1%–18.1%) but significant variation in host taxonomy (4.2%–65.3%), with copiotrophs showing higher values than oligotrophs. The high infection rates of copiotrophs were attributed to collective infection by diverse viruses, suggesting weak density-dependent virus–host selection, presumably due to their nonpersistent interactions with viruses resulting from fluctuating abundance. In contrast, the low infection rates of oligotrophs supported the idea that their codominance with viruses is achieved by genomic microdiversification, which diversifies the virus–host specificity, sustained by their large population size and persistent density-dependent fluctuating selection. Notably, we discovered viruses infecting CL500-11, the dominant bacterioplankton lineage in deep freshwater lakes worldwide. These viruses showed extremely high read coverages in cellular and virion metagenomes but were detected in <1% of host cells, suggesting a low infection rate and high burst size. Overall, we revealed highly diverse virus–host interactions within and between host lineages that were overlooked at the metagenomic resolution.
|
|
Scooped by
?
Today, 2:21 PM
|
Halomonas species have recently emerged as promising chassis organisms for next-generation industrial biotechnology, due to their ability to thrive under high-salt conditions, where most microorganisms cannot survive. This feature minimizes contamination risks, thus enabling cultivation under open, unsterile conditions. In addition, many Halomonas species naturally produce large amounts of the bioplastic polyhydroxybutyrate PHB and the high-value osmolyte ectoine. This review explores the development of genetic manipulation tools and their pivotal role in establishing the genus Halomonas as an industrial chassis. Key additions to the synthetic biology toolbox, including cloning vectors, genetic parts, and genome editing systems are highlighted, along with challenges faced for their adoption, such as difficulties in transformation. In addition, we showcase how these tools have been employed for the development of more robust, high-producing strains through metabolic engineering, as well as for expanding the portfolio of target metabolites produced by Halomonas. Recent developments in synthetic biology tools and metabolic engineering highlighted in this review underscore the potential of Halomonas for large scale metabolite production and provide a promising outlook towards their role as a microbial chassis in industrial biotechnology.
|
|
Scooped by
?
Today, 2:11 PM
|
Virus-like particles (VLPs) are self-assembling nanoparticles derived from viruses with potential as scaffolds for myriad applications. They are also excellent testbeds for engineering protein superstructures. Engineers often employ techniques such as amino acid substitutions and insertions/deletions. Yet evolution also utilizes circular permutation, a powerful natural strategy that has not been fully explored in engineering self-assembling protein nanoparticles. Here, we demonstrate this technique using the MS2 VLP as a model self-assembling, proteinaceous nanoparticle. We constructed, for the first time, a comprehensive circular permutation library of the fused MS2 coat protein dimer construct. The strategy revealed new terminal locations, validated via cryo-electron microscopy, that enabled C-terminal peptide tagging and led to a stable protein encapsulation strategy via covalent bonding - a feature the native coat protein does not permit. Our systematic study demonstrates the power of circular permutation for engineering new features as well as quantitatively and systematically exploring VLP structural determinants.
|
|
Scooped by
?
Today, 1:05 AM
|
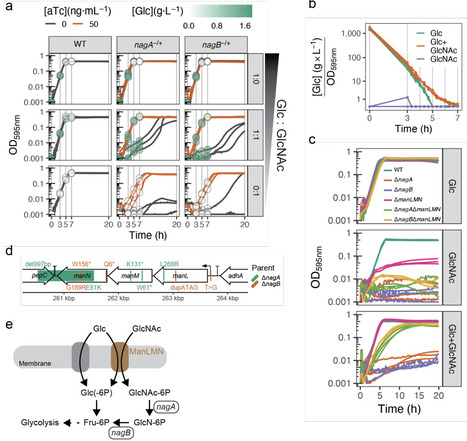
The human body comprises many different microenvironments, each with their own challenges for microorganisms to overcome in order to survive, and possibly cause infection. The human pathogen Streptococcus pneumoniae is notoriously flexible in this regard, and can adapt to a wide range of host niches, including the nasopharynx, lungs, and cerebrospinal fluid. However, the molecular and genetic underpinnings of this ability remain largely obscure. In this work, using infection-mimicking growth conditions we demonstrate that niche adaptation imposes genome-wide changes on multiple levels, including gene essentiality, expression and membrane lipid composition. In general, we show that gene expression and fitness profiling couple orthogonal sets of genes to environmental stimuli. For instance, N-acetylglucosamine (GlcNAc) import (manLMN) and catabolism (nagAB) genes were required for growth on this sugar, but not differentially expressed in its presence, whereas other amino sugar metabolism pathways were upregulated, but not essential. Surprisingly, we found that pneumococci do not necessarily prefer glucose over GlcNAc and that uptake of GlcNAc in absence of subsequent catabolism was toxic. Moreover, we identified a previously overlooked fatty acid saturation regulator, FasR, controlling membrane composition, rendering it important during heat stress. A fundamental understanding of how genes contribute to bacterial niche adaptation, including nutrient availability or temperature fluctuations, is crucial for understanding successful antibiotic therapy and vaccination strategies and the development of novel anti-infectives.
|
|
Scooped by
?
Today, 12:52 AM
|
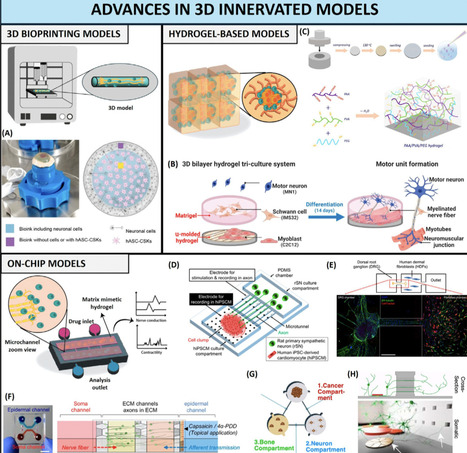
Innervation plays a key role in tissue homeostasis, disease, and repair throughout our lifetimes. Strategies for emulating innervation and its bioinstructive influence on surrounding cells are thus highly desirable to upgrade the organotypic features of human in vitro models. We delve into the latest strategies for generating innervated 3D models that mimic native innervation patterns, and highlight recent advances in bioengineering living platforms for emulating the effect of innervation during tissue regeneration or for recapitulating tumor–nerve interplay. We present an overview of the key design blueprints for generating innervated tissue models, highlight the challenges to be overcome, and discuss the biomedical breakthroughs that can branch from such in vitro platforms.
|
|
Scooped by
?
Today, 12:37 AM
|
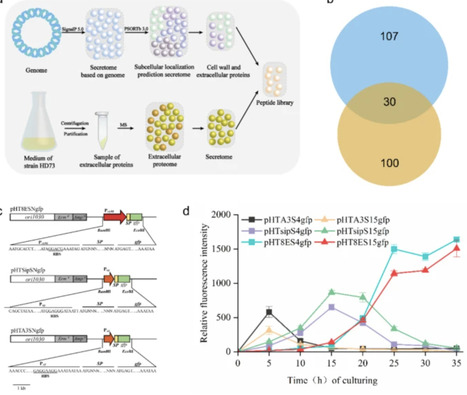
Efficient secretion of heterologous proteins is crucial for applications in industrial and biomedical fields. Selecting appropriate signal peptides and bacterial strains is critical for successful protein expression and export. Bacillus thuringiensis, known for its robust secretion capabilities within the Bacillus genus, shows promise as an ideal host for this purpose. We performed genome-based bioinformatic analysis of B. thuringiensis HD73. A total of 525 proteins were predicted to contain signal peptides, exceeding those in other Bacillus species. The extracellular proteome of B. thuringiensis HD73 was analyzed via LC-MS/MS, identifying 100 secreted proteins. A library of 30 signal peptides was constructed by integrating genome-based predictions with experimental secretome data. Using this library, green fluorescent protein secretory expression systems were developed in the acrystalliferous mutant strain B. thuringiensis HD73−, and the strain carrying signal peptide S17 showed the highest secretion efficiency. Additionally, the top 10 performing signal peptides were used to express and secrete the convenient enzyme cutinase, with the S20 fusion strain exhibiting the highest cutinase activity (3.65 U/mL in the culture supernatant). This study provides the first combined bioinformatic and experimental characterization of the B. thuringiensis secretome. The developed secreted protein expression system and signal peptide library demonstrate effectiveness and offer potential for future heterologous protein secretion in B. thuringiensis.
|
|
Scooped by
?
Today, 12:06 AM
|
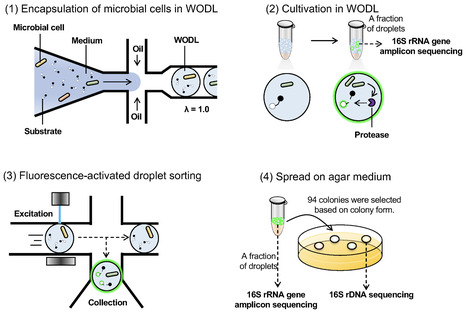
Exploration of diverse microbial sources, particularly environmental bacteria, is needed to identify novel and efficient peptidases/proteases that can be used in a variety of industrial applications. However, conventional function-based screening methods are inefficient and preclude the use of diverse microbial resources. This study illustrates a revolutionary approach to microbial screening using droplet-based microfluidics and fluorescence-activated droplet sorting that targets endopeptidases. Droplet-based microfluidic systems are a powerful tool for culturing microorganisms and for detecting microbial functions inside droplets with ultra-high throughput. However, droplet-based microfluidics for screening the proteolytic activity of environmental bacteria at a large scale remains largely unexplored. Here, we screened approximately 630,000 microorganisms in 6 h and obtained four species with high peptidase activity using droplet-based microfluidics. Furthermore, we isolated an Asp-specific endopeptidase from the isolated bacteria Lysobacter soli and showed that its activity was 2.4-fold higher than that of the related commercially available enzyme. The successful isolation of Asp-specific peptidase with superior activity in a short period of time compared to existing alternatives underscores the efficacy of droplet-based microfluidics for function-based microbial screening.
|
|
Scooped by
?
June 14, 11:46 PM
|
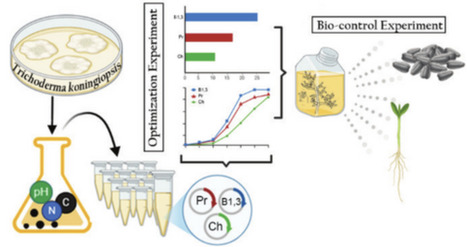
The growing demand for sustainable alternatives to chemical fungicides has driven the development of microbial-based biocontrol strategies. In this study, the native strain Trichoderma koningiopsis LBM116 (Misiones, Argentina) was optimized for the production of mycolytic enzymes (chitinases, β-1,3-glucanases, and proteases) using factorial and response surface experimental designs. Enzyme secretion was increased by more than 250% compared to initial conditions by selecting specific carbon and nitrogen sources and adjusting inoculum and pH parameters. The optimized enzyme formulation improved lettuce seed germination to 86.66% in the presence of the phytopathogen Fusarium sp., under controlled conditions. In seedling trials, it also reduced disease severity and improved growth parameters. These results confirm the dual effect of the enzyme formulation, acting as a biocontrol agent and plant growth promoter. This work highlights the potential of enzyme formulations derived from T. koningiopsis LBM116 as an effective, low-cost, and sustainable alternative for managing phytopathogens in agriculture.
|
|
Scooped by
?
June 14, 11:15 PM
|
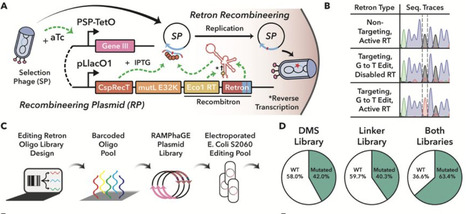
Allostery, the transmission of locally induced conformational changes to distant functional sites, is a key mechanism for protein regulation. Artificial allosteric effectors enable remote manipulation of cell function; their engineering, however, is hampered by our limited understanding of allosteric residue networks. Here, we introduce a phage-assisted evolution platform for in vivo optimization of allosteric proteins. It applies opposing selection pressures to enhance activity and switchability of phage-encoded effectors and leverages retron-based recombineering to broadly explore fitness landscapes, covering point mutations, insertions and deletions. Applying our pipeline to the transcription factor AraC yielded optogenetic variants with light-controlled activity spanning ∼1000-fold dynamic range. Long-read sequencing across selection cycles revealed adaptive trajectories and corresponding allosteric interactions. Our work facilitates phage-assisted evolution of allosteric proteins for programmable cellular control.
|
|
Scooped by
?
June 13, 4:51 PM
|
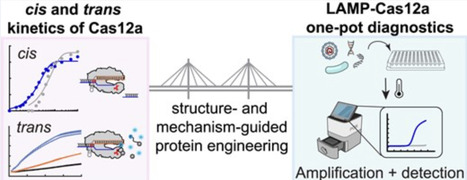
Cas12a trans nuclease activity has been leveraged for nucleic acid detection, often coupled with isothermal amplification to increase sensitivity. However, due to the lack of highly efficient thermostable Cas12a orthologs, use of Cas12a in one-pot combination with high temperature (55–65°C) amplification, such as loop-mediated isothermal amplification (LAMP), has remained challenging. Here, we attempt to address this challenge by comparative study of the thermostable, but poorly trans-active YmeCas12a (from Yellowstone metagenome) with the mesophilic, highly trans-active LbaCas12a (from Lachnospiraceae bacterium ND 2006). Kinetic characterizations identified that poor trans substrate affinity (high Km) is the key limiting factor in YmeCas12a trans activity. We engineered YmeCas12a by structure-guided mutagenesis and fusion to DNA-binding domains to increase its affinity to the trans substrate. The most successful combinatorial variant showed 5–50-fold higher catalytic efficiency with the trans substrate depending on the target site. With the improved variant, we demonstrate efficient nucleic acid detection in combination with LAMP in a single reaction workflow, setting the basis for development of point-of-care tests.
|
|
Scooped by
?
June 13, 4:33 PM
|
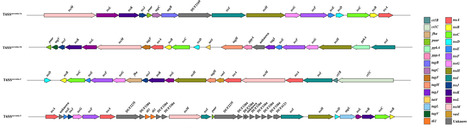
The type VI secretion system (T6SS) is a contact-dependent contractile nanomachine widely distributed among Gram-negative bacteria, essential for interbacterial competition, host interactions and environmental adaptation. The T6SS encoded by Serratia is known to play roles in bacterial competition and contribute to niche adaptation. Yet, the diversity and distribution of T6SS loci and relevant proteins across different Serratia species remain underexplored. In this study, we conducted bioinformatic and comparative genomic analyses of the T6SS to expand our understanding of T6SS in Serratia and their distribution within the genus. A dataset comprising 2,337 Serratia genomes was analysed, identifying 4 distinct T6SS loci classified into 3 main types based on TssB sequence variation, with varying distributions across specific Serratia haplogroups. T6SSserratia-1b subtype was found to be the most prevalent. By integrating homology searches and positional information, we expanded the known database of T6SS proteins in Serratia, identifying a range of effectors likely involved in interbacterial interactions and host adaptation. Additionally, statistical analysis reveals a strong correlation between the presence of T6SS and the diversity of associated genes, including not only known T6SS effectors and immunity proteins but also other co-occurring gene families, enabling the identification of multiple candidate loci potentially involved in bacterial competition and pathogenicity. These findings shed light on the complex distribution, evolutionary dynamics and functional diversity of T6SS and associated proteins in Serratia, advancing our understanding of their role in bacterial competition and environmental adaptation.
|
|
|
Scooped by
?
Today, 2:56 PM
|
Ultrasensitive transcriptional switches are essential for converting gradual molecular inputs into decisive gene expression responses, enabling critical behaviors such as bistability and oscillations. While cooperative binding, relying on direct repressor-DNA binding, has been classically regarded as a key ultrasensitivity mechanism, recent theoretical works have demonstrated that combinations of indirect repression mechanisms—sequestration, blocking, and displacement—can also achieve ultrasensitive switches with greater robustness to transcriptional noise. However, these previous works have neglected key biological constraints such as DNA binding kinetics and the limited availability of transcriptional activators, raising the question of whether ultrasensitivity and noise robustness can be sustained under biologically realistic conditions. Here, we systematically assess the impact of these factors on ultrasensitivity and noise robustness under physiologically plausible conditions. We show that while various repression combinations can reduce noise, only the full combination of all three indirect mechanisms consistently maintains low noise and high ultrasensitivity. As a result, biological oscillators employing this triple repression architecture retain precise rhythmic switching even under high noise, and even when activators are shared across thousands of target genes. Our findings offer a mechanistic explanation for the frequent co-occurrence of these repression mechanisms in natural gene regulatory systems.
|
|
Scooped by
?
Today, 2:40 PM
|
Cis regulatory elements (CREs) interact with their target promoters over long genomic distances and can be identified using chromatin conformation capture (3C) assays. Their regulatory activity can be functionally characterized in a high-throughput manner using massively parallel reporter assays (MPRAs) that generally test an enhancer alongside a minimal promoter. Here, we developed a novel technology called Capture-C MPRA (ccMPRA) that combines both technologies and can simultaneously obtain chromatin interactions and measure CRE activity alongside their target promoters. We utilized ccMPRA to analyze the regulatory activity of 650 promoters interacting with 42,719 sequences. As C-based techniques also capture isolated promoters, we were able to obtain promoter baseline activity, enabling the identification of both enhancers and silencers. Analysis of CREs interacting with more than one promoter showed significant activity differences depending on the promoter. In summary, ccMPRA can simultaneously characterize chromatin interactions and regulatory activity, allowing to further dissect regulatory grammar.
|
|
Scooped by
?
Today, 2:26 PM
|
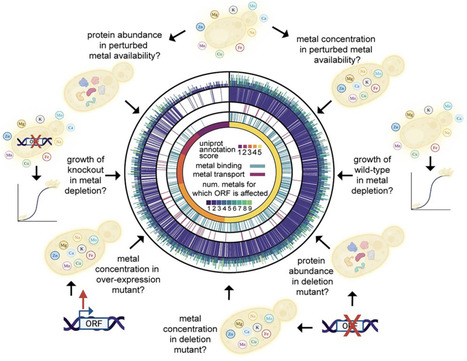
Metal ions have crucial roles in cells, but the impact of their availability on biological networks is underexplored. We systematically quantified yeast cell growth and the corresponding metallomic, proteomic, and genetic responses to perturbations in metal availability along concentration gradients of all growth-essential metal ions. We report a remarkable metal concentration dependency of cellular networks, with around half of the proteome, and most signaling pathways such as target of rapamycin (TOR), being metal responsive. Although the biological response to each metal is distinct, our data reveal common properties of metal responsiveness, such as concentration interdependencies and metal homeostasis. Furthermore, our resource indicates that many understudied proteins have functions related to metal biology and reveals that metalloenzymes occupy central nodes in metabolic networks. This work provides a framework for understanding the critical role of metal ions in cellular function, with broader implications for manipulating metal homeostasis in biotechnology and medicine.
|
|
Scooped by
?
Today, 2:14 PM
|
Plasmid DNA (pDNA) is a cost-driving reagent for the production of gene therapies and DNA vaccines. Improving pDNA production in the most common production host (E. coli) has faced obstacles arising from the complex network of genes responsible for pDNA synthesis, with the specific enzyme(s) limiting pDNA yield remaining unidentified. To address this challenge, we employed an inducible genome-wide mutagenesis strategy, combined with fluorescent screening, to isolate E. coli NEB 5α strains with enhanced pDNA production. Following selection, we successfully isolated an E. coli strain (M3) with elevated plasmid copy numbers (PCNs) across multiple origin types. Specifically, we observed a 5.93-fold increase in PCN for the GFP reporter plasmid, a 1.93-fold increase for the gWiz DNA vaccine plasmid, and an 8.7-fold increase for the pAAV-CAGG-eGFP plasmid, all of which contain pUC origins. In addition, plasmids with p15A and pSC101 origins showed 1.44-fold and 1.68-fold increases in PCN, respectively. Whole-genome sequencing of the adapted strain M3 identified 85 mutations, including one in recG, which encodes an ATP-dependent DNA helicase. Replacement of the mutant recG with its wild-type counterpart in the mutant strain resulted in a 63% reduction in PCN, but the recG mutation alone was insufficient to increase PCN in the wild-type strain. These findings suggest that the recG mutation plays a synergistic role with other genomic mutations to drive PCN increases. Taken together, this study presents the development of a pDNA hyperaccumulating E. coli strain with promising applications in industrial and therapeutic pDNA production, while also offering important insights into key genes involved in pDNA production.
|
|
Scooped by
?
Today, 1:08 AM
|
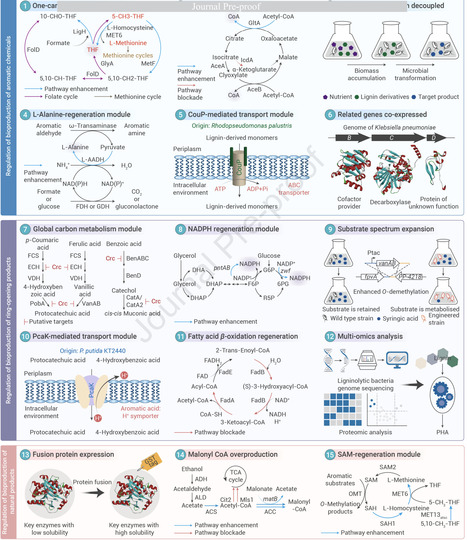
Lignin, a natural and renewable aromatic polymer, serves as a plentiful source of aromatic building blocks for biomanufacturing. However, despite its potential, the bioconversion efficiency of lignin remains limited due to its inherently complex structure and the constraints of traditional metabolic engineering approaches. Synthetic biology-guided metabolic regulation offers a solution by coordinating intracellular resource allocation in ligninolytic strains, endowing their adaptation for industrial applications and promoting the sustainability of lignin-based bioeconomy. This review provides a comprehensive overview of multiscale metabolic regulation, including critical enzymes involved in biotransformation, metabolic pathway networks, genome-phenotype and learning prediction. It highlights the significant roles and potential benefits of emerging cutting-edge technologies in advancing lignin valorization. Overall, synthetic biology-guided metabolic regulation has demonstrated its power in balancing the metabolic fluxes of ligninolytic strains, ensuring the continued vitality in future development.
|
|
Scooped by
?
Today, 12:56 AM
|
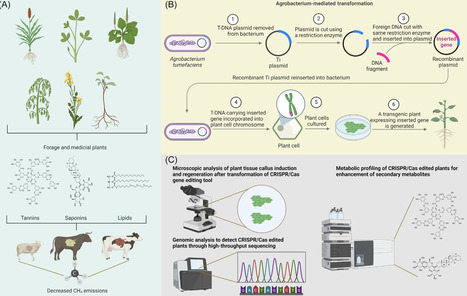
Methane emissions from ruminants represent a major contributor to global greenhouse gases (GHGs), posing challenges for sustainable agriculture and climate change mitigation. Recent advances in CRISPR-Cas-based gene editing offer transformative approaches to address this issue by targeting both forage crops and rumen methanogens. Enhancing lipid content and secondary metabolites in forage crops can suppress methanogenesis and improve feed efficiency, while precise genome editing in methanogenic Archaea can disrupt pathways critical to methane production. However, challenges remain regarding delivery methods, gene targeting specificity, ecological impacts, and regulatory acceptance. In this review, we explore current progress, identify key knowledge gaps, and highlight the need for interdisciplinary strategies that integrate biotechnology, synthetic biology, and regulatory frameworks to develop effective and scalable methane mitigation solutions.
|
|
Scooped by
?
Today, 12:40 AM
|
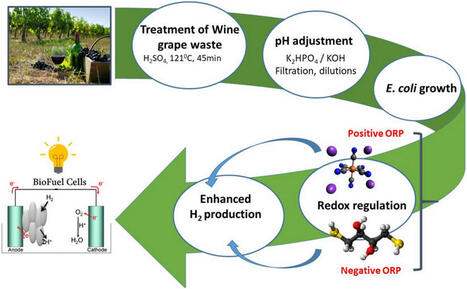
Lignocellulosic wine grape waste (WGW) is a cheap medium for Escherichia coli growth and H2 production. The current study investigated the effect of initial redox potential (oxidation–reduction potential (ORP)) on the growth, H2 production, ORP kinetics, and current generation of the E. coli BW25113 parental and a mutant strain optimized for hydrogen evolution when fermenting WGW (40 g L−1) hydrolysate. Bacteria were cultivated anaerobically on pre-treated WGW hydrolysate with dilutions ranging from undiluted to fourfold dilution, at pH 7.5. Notably, a twofold diluted medium, with pH adjustment using K2HPO4, exhibited reduced acidification, prolonged H2 production, and enhanced biomass formation (OD600, 1.5). The addition of the redox reagent DL-dithiothreitol (DTT) was found to positively influence the H2 production of both the E. coli BW25113 parental and mutant strains. H2 production started after 24 h of growth, reaching a maximum yield of 5.10 ± 0.02 mmol/L in the wild type and 5.3 ± 0.02 mmol/L in the septuple mutant strain, persisting until the end of the stationary growth phase. The introduction of 3 mM DTT induced H2 production from the early-exponential phase, indicating that reducing conditions enhanced H2 production. Furthermore, we assessed the efficacy of using intact E. coli cells (1.5 mg cell dry weight) as anode catalyst in a bio-electrochemical fuel-cell system. Whole cells of the septuple mutant grown under reduced ORP conditions yielded the highest electrical potential, reaching up to 0.7 V. The results highlight the potential of modifying medium buffering capacity and ORP as a tool to improve biomass yield and H2 production during growth on WGW for biotechnological biocatalyst applications.
|
|
Scooped by
?
Today, 12:30 AM
|
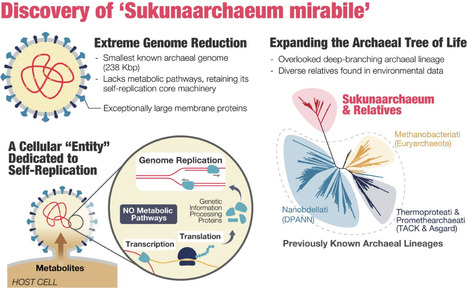
Defining the minimal genetic requirements for cellular life remains a fundamental question in biology. Genomic exploration continually reveals novel microbial lineages, often exhibiting extreme genome reduction, particularly within symbiotic relationships. Here, we report the discovery of Candidatus Sukunaarchaeum mirabile, a novel archaeon with an unprecedentedly small genome of only 238 kbp —less than half the size of the smallest previously known archaeal genome— from a dinoflagellate-associated microbial community. Phylogenetic analyses place Sukunaarchaeum as a deeply branching lineage within the tree of Archaea, representing a novel major branch distinct from established phyla. Environmental sequence data indicate that sequences closely related to Sukunaarchaeum form a diverse and previously overlooked clade in microbial surveys. Its genome is profoundly stripped-down, lacking virtually all recognizable metabolic pathways, and primarily encoding the machinery for its replicative core: DNA replication, transcription, and translation. This suggests an unprecedented level of metabolic dependence on a host, a condition that challenges the functional distinctions between minimal cellular life and viruses. The discovery of Sukunaarchaeum pushes the conventional boundaries of cellular life and highlights the vast unexplored biological novelty within microbial interactions, suggesting that further exploration of symbiotic systems may reveal even more extraordinary life forms, reshaping our understanding of cellular evolution.
|
|
Scooped by
?
June 14, 11:55 PM
|
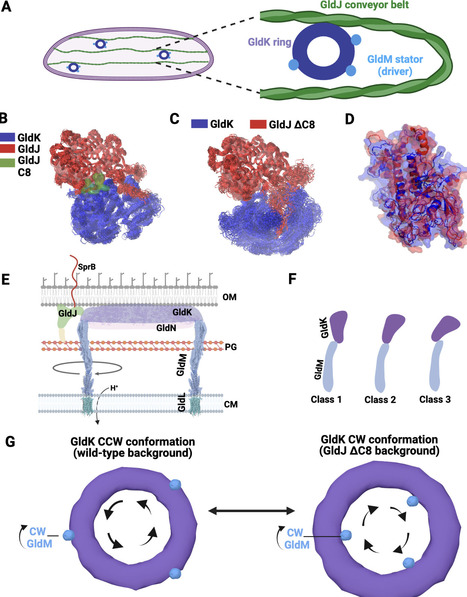
Many bacteria utilize the type 9 secretion system (T9SS) for gliding motility, surface colonization, and pathogenesis. This dual-function motor supports both gliding motility and protein secretion, where rotation of the T9SS plays a central role. Fueled by the energy of the stored proton motive force and transmitted through the torque of membrane-anchored stator units, the rotary T9SS propels an adhesin-coated conveyor belt along the bacterial outer membrane like a molecular snowmobile, thereby enabling gliding motion. However, the mechanisms controlling the rotational direction and gliding motility of T9SS remain elusive. Shedding light on this mechanism, we find that in the gliding bacterium Flavobacterium johnsoniae, deletion of the C-terminus of the conveyor belt-associated protein GldJ controls and, in fact, reverses the rotational direction of T9SS from counterclockwise (CCW) to clockwise (CW). This suggests that the interface between the conveyor belt-associated protein GldJ and the T9SS ring protein GldK plays an important role in controlling the directionality of T9SS, potentially by modulating its interaction with the stator complex GldLM, which drives motor rotation. Combined with MD simulation of the T9SS stator units GldLM, we suggest a “tri-component gearset” model where GldJ controls the rotational direction of its driver, the T9SS, thus providing adaptive sensory feedback to influence the motility of the gliding bacterium.
|
|
Scooped by
?
June 14, 11:38 PM
|
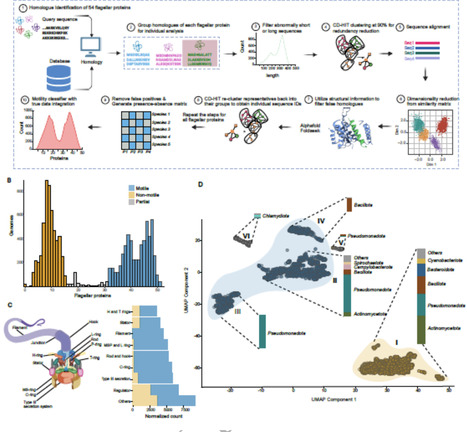
Bacterial swimming is mostly powered by the bacterial flagellar motor and the number of proteins involved in the flagellar motor can vary. Quantifying the proteins present in flagellar motors from a range of species delivers insight into how motility has changed throughout history and provides a platform for estimating from its genome whether a species is likely to be motile. We conducted sequence and structural homology searches for 54 flagellar pathway proteins across 11 365 bacterial genomes and developed a classifier with up to 95% accuracy that could predict whether a strain was motile or not. We then mapped the evolution of flagellar motility across the GTDB bacterial tree of life. We confirmed that the last common bacterial ancestor had flagellar motility and that the rate of loss of this motility was four-fold higher than the rate of gain. We showed that the presence of filament protein homologues was highly phylogenetically correlated with motility and that all species classified as motile contained at least one filament homologue. We calculated the rate of gain and loss for each flagellar protein and that the filament protein FliC was highly correlated with motility across the tree of life. We then measured the correlation of each flagellar motor protein with FliC and showed that the filament, rotor, and rod and hook proteins were all highly correlated with FliC, and thus with motility. We calculated the differential rates of gain and loss for each flagellar protein and quantified which genomes encoded for partial sets of flagellar proteins, indicating potential pathways by which motility could be lost. Overall, we show that filament, rod and hook and rotor proteins are conserved when flagellar motility is preserved and that the presence or absence of a FliC homologue is a good, simple predictor of whether or not a species has flagellar motility.
|
|
Scooped by
?
June 13, 4:54 PM
|
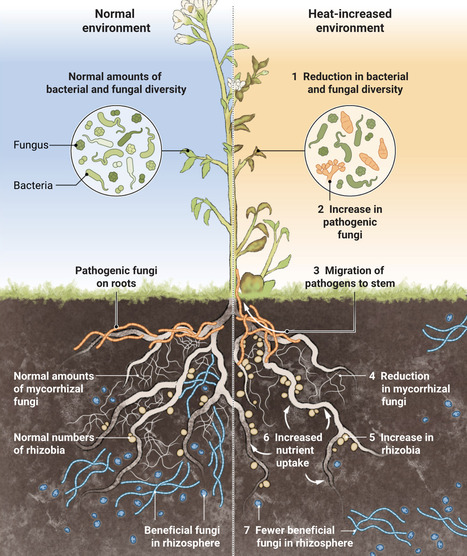
Rising temperatures change the structure and function of plant microbial communities.
|
|
Scooped by
?
June 13, 4:46 PM
|
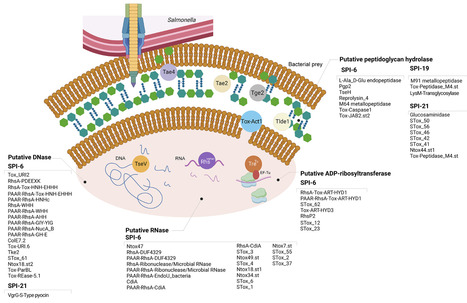
The Type VI Secretion System (T6SS) is a critical fitness and virulence factor of many Gram-negative bacteria. Five T6SS gene clusters have been described in Salmonella, each one encoded within different pathogenicity islands (i.e., SPI-6, SPI-19, SPI-20, SPI-21, and SPI-22). The events of gain and loss of these T6SS gene clusters have contributed to shape the genome evolution of different Salmonella serotypes. In addition, the differential distribution of T6SS and the ever-increasing repertoire of predicted effector proteins is likely to play an important role in the environmental fitness and pathogenic potential of different Salmonella serotypes. This review summarizes the current knowledge on the role played by T6SS in Salmonella biology, highlighting the major milestones in the field over the past two decades. We discuss the expanding repertoire of T6SS effector proteins identified to date and examine the current understanding of mechanisms controlling T6SS expression in Salmonella, focusing on host-derived cues and regulators involved. Finally, we provide a critical analysis of conflicting reports and suggest future directions for the research in the field. A better understanding of these processes could expand our knowledge of Salmonella biology, and the mechanisms behind how this versatile secretion system enables Salmonella to thrive in competitive microbial environments and contribute to host colonization.
|