 Your new post is loading...

|
Scooped by
?
Today, 1:37 AM
|
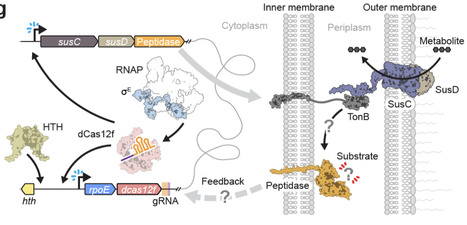
Bacterial transcription initiation is a tightly regulated process that canonically relies on sequence-specific promoter recognition by dedicated sigma (σ) factors, leading to functional DNA engagement by RNA polymerase (RNAP). Although the seven σ factors in E. coli have been extensively characterized, Bacteroidetes species encode dozens of specialized, extracytoplasmic function σ factors (σE) whose precise roles are unknown, pointing to additional layers of regulatory potential. Here we uncover an unprecedented mechanism of RNA-guided gene activation involving the coordinated action of σE factor in complex with nuclease-dead Cas12f (dCas12f). We screened a large set of genetically-linked dCas12f and σE homologs in E. coli using RIP-seq and ChIP-seq experiments, revealing systems that exhibited robust guide RNA enrichment and DNA target binding with a minimal 5ʹ-G target-adjacent motif (TAM). Recruitment of σE was dependent on dCas12f and guide RNA (gRNA), suggesting direct protein-protein interactions, and co-expression experiments demonstrated that the dCas12f-gRNA-σE ternary complex was competent for programmable recruitment of the RNAP holoenzyme. Remarkably, dCas12f-RNA-σE complexes drove potent gene expression in the absence of any requisite promoter motifs, with de novo transcription start sites defined exclusively by the relative distance from the dCas12f-mediated R-loop. Our findings highlight a new paradigm of RNA-guided transcription (RGT) that embodies natural features reminiscent of CRISPRa technology developed by humans.
|
|
Scooped by
?
Today, 1:11 AM
|
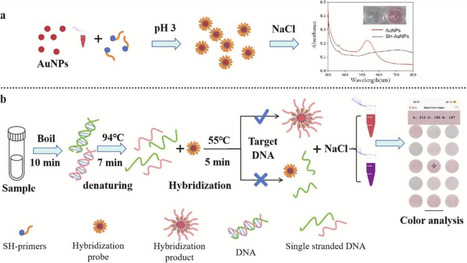
Total bacterial counts (TBC) and coliforms bacteria have been used as the index of microbial quality and fecal contamination for foods, the detection of them is involved in microbiological criteria of most countries to guarantee food safety. In this study, a gold nanoparticle (AuNPs) based colorimetric assay for rapid detection of TBC and coliform bacteria was developed by using the molecular hybridization approach. Thiol modified primers SH-27F and SH-lacZF were designed and conjugated with AuNPs, which was acted as both hybridization probe and signal probe, and salt induced aggregation of AuNPs was indicator of the hybridization and detection. Escherichia coli was studied as model strain in this study. The result showed that under the optimal conditions, the limit of detection for TBC and coliforms bacteria were 102 CFU/mL and 101 CFU/mL by visual detection, and 101 CFU/mL by UV–vis spectrum analysis, a smart phone application named ColorPicker was used for the quantitative analysis of the colorimetric assay. The color signal had good linear relationship of E. coli concentration in the range of 101–104 CFU/mL. The detection is rapid, simple and sensitive, without the needing of culture enrichment and precious equipment, has good application potential for the on-site detection of TBC and coliforms bacteria. In addition, the proposed strategy provides a direction for the design of highly sensitive colorimetric signal probes for other pathogenic microorganisms.
|
|
Scooped by
?
Today, 1:02 AM
|
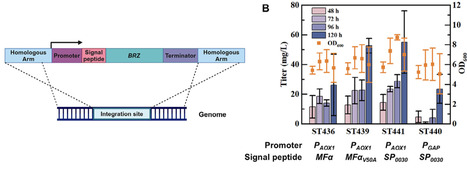
Excessive sugar consumption is associated with metabolic disorders such as obesity and diabetes, prompting the search for healthier alternatives. Sweet-tasting proteins offer high sweetness, safety, and low caloric content, making them promising sugar substitutes. However, their application is hindered by limited natural abundance and challenging extraction processes from plant sources. In this study, the microorganism-based sweet-tasting protein was explored. Brazzein was first verified as an expressible protein for production using the Komagataella phaffii expression system. Through the evaluation of promoters and signal peptides, the PAOX1→SP0030→PbBRZ→Tdas1 expression cassette was identified as optimal for brazzein production. Via the optimization of brazzein gene copy number and the modulation of protein translation, folding, and degradation processes, brazzein production was further improved to 113.24 and 166.54 mg/L, respectively. By performing fed-batch fermentation in a 5 L bioreactor, the engineered strain produced brazzein at a titer of 639.11 mg/L. This is the highest level for microbial brazzein production. These findings highlight the effectiveness of our strain optimization strategies and provide a foundation for the industrial application of sweet-tasting proteins.
|
|
Scooped by
?
Today, 12:48 AM
|
Cupriavidus necator is a promising bacterium for producing polyhydroxybutyrate (PHB), a biodegradable bioplastic. However, the cell growth is restricted due to a lack of glucose transporters and low glucokinase activity. To address this limitation, we first developed comprehensive genetic toolkits to express sfGFP as a proof-of-concept in C. necator. A plasmid-driven T7 RNA polymerase (PDT7) system under the J23109 promoter achieved a 10-fold increase in fluorescence compared to strains without PDT7. However, PDT7 imposed a metabolic burden at higher expression levels and remained less effective than the constitutive Trc promoter, which consistently exhibited the highest transcriptional strength. Based on its robust and balanced performance, the Trc promoter was optimized to drive expression of galP (galactose permease) and glk (glucokinase) genes from Escherichia coli, (annotated as Tgg-H16), enabling enhanced glucose uptake, biomass and PHB biosynthesis. Further enhancement was achieved by supplementing a mixed carbon source, that is, 10 g/L glucose and 10 g/L fructose, which shortened the lag phase and supported higher PHB yields. Finally, a stepwise feeding strategy in fermentation boosted PHB production to 30.9 g/L. Rewiring carbon flux in C. necator through genetic circuit design demonstrates the feasibility of improving carbon uptake, while integration with tailored cultivation strategies enables upcycling sustainable PHB production.
|
|
Scooped by
?
June 10, 11:51 PM
|
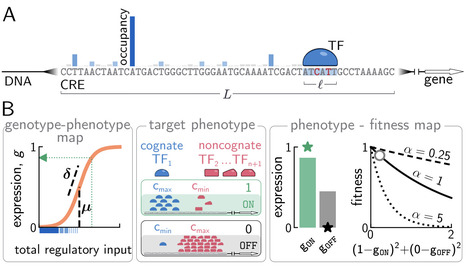
Cis-regulatory elements (CREs), such as enhancers and promoters, control gene expression by binding regulatory proteins. In contrast to their bacterial counterparts, eukaryotic CREs typically bind transcription factors with short recognition motifs across multiple functional yet often weak binding sites. The evolutionary origin of this genetic architecture remains unclear. Here we study adaptive evolution of entire CREs under selection for controllable gene expression. In a biophysical toy model that recapitulates the essential nonlinearities of eukaryotic regulation, a regulatory phenotype requires a gene to be active when its CRE binds cognate TFs, yet inactive in the presence of noncognate TFs that can cause deleterious crosstalk. We explore CRE evolutionary outcomes utilizing "optimize-to-adapt" approach suggested by the theory presented in a companion paper. In this approach, CRE evolution is simulated explicitly at the sequence level, while the parameters of the biophysical model itself -- i.e., the properties of the replicated genotype-phenotype map -- are numerically optimized for CRE evolvability. In the optimal regime, selection navigates the tradeoff between slowly evolving strong and long binding sites (which guarantee low crosstalk), and rapidly evolving multiple short and weak binding sites (which necessitate diffuse selection against excessive noncognate binding across the entire CRE). When we further explore various scenarios for cooperative regulation, we find that the optimal regime predicts a diversity of strong and weak, yet always short, binding sites and favors "synergistic activation" of transcription, as reported empirically for eukaryotes. These results showcase how information theory can link evolutionary dynamics with biophysical constraints to rationalize -- and possibly even predict -- optimal regulatory architectures.
|
|
Scooped by
?
June 10, 11:24 PM
|
Accumulating evidence indicates that microorganisms respond to the ubiquitous plastic pollution by evolving plastic-degrading enzymes. However, the functional diversity of these enzymes and their distribution across the ocean, including the deep sea, remain poorly understood. By integrating bioinformatics and artificial intelligence-based structure prediction, we developed a structure- and function–informed algorithm to computationally distinguish functional polyethylene terephthalate-degrading enzymes (PETases) from variants lacking PETase activity (pseudo-PETase), either due to alternative substrate specificity or pseudogene origin. Through in vitro functional screening and in vivo microcosm experiments, we verified that this algorithm identified a high-confidence, searchable sequence motif for functional PETases capable of degrading PET. Metagenomic analysis of 415 ocean samples revealed 23 PETase variants, detected in nearly 80% of the samples. These PETases mainly occur between 1000 and 2000 m deep and at the surface in regions with high plastic pollution. Metatranscriptomic analysis further identified PETase variants that were actively transcribed by marine microorganisms. In contrast to their terrestrial counterparts—where PETases are taxonomically diverse—those in marine ecosystems were predominantly encoded and transcribed by members of the Pseudomonadales order. Our study underscores the widespread distribution of PETase-containing bacteria across carbon-limited marine ecosystems, identifying and distinguishing the PETase motif that underpins the functionality of these specialised cutinases.
|
|
Scooped by
?
June 10, 11:09 PM
|
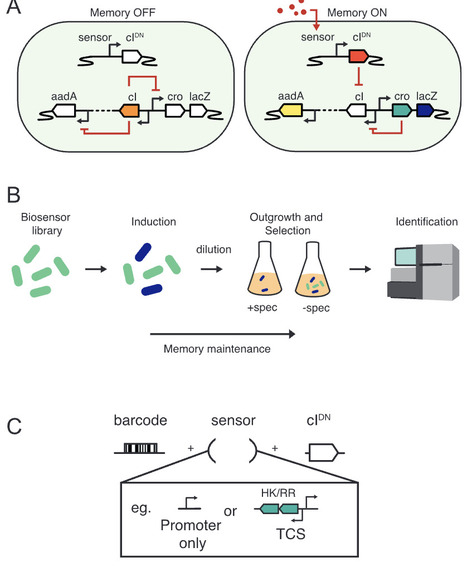
Synthetic biology approaches such as whole-cell biosensing and ‘sense-and-respond’ therapeutics aim to enlist the vast sensing repertoire of gut microbes to drive cutting-edge clinical and research applications. However, well-characterised circuit components that sense health- and disease-relevant conditions within the gut remain limited. Here, we extend the flexibility and power of a biosensor screening platform using bacterial memory circuits. We construct libraries of sensory components sourced from diverse gut bacteria using a bespoke two-component system identification and cloning pipeline. Tagging unique strains using a hypervariable DNA barcode enables parallel tracking of thousands of unique clones, corresponding to ~150 putative biosensors, in a single experiment. Evaluating sensor activity and performance heterogeneity across various in vitro and in vivo conditions using mouse models, we identify several biosensors of interest. Validated hits include biosensors with relevance for autonomous control of synthetic functions within the mammalian gut and for non-invasive monitoring of inflammatory disease using faecal sampling. This approach will promote rapid biosensor engineering to advance the development of synthetic biology tools for deployment within complex environments.
|
|
Scooped by
?
June 10, 10:35 PM
|
Biofilm formation is the most effective pathway for electron transfer to anodes in bioelectrochemical systems. However, the mechanisms triggering biofilm formation under anoxic conditions, as well as the architectural and compositional factors that positively influence current generation, are not well understood. Recent findings have shown that riboflavin can function similarly to a quorum sensing molecule in the γ-proteobacterium Shewanella oneidensis. Enhanced biofilm formation induced by riboflavin correlates with increased current densities. Only a limited number of candidate proteins were found to have altered concentrations due to this quorum sensing mechanism. This study demonstrates that the catalytic functions of the UDP-N-acetylglucosamine C4 epimerase WbpP and UDP-N-acetyl-D-glucosamine 6-dehydrogenase WbpA affect biofilm formation and lead to increased current density. Using optical coherence tomography, we found that the expression of each protein individually causes specific, quantifiable changes in biofilm architecture, including biovolume, height, and porosity. However, the current density did not significantly differ when these proteins were expressed alone compared to the control. In contrast, co-expression of WbpP and WbpA resulted in a doubling of current density, closely resembling the increases observed with riboflavin-mediated quorum sensing. We hypothesize that riboflavin-based quorum sensing may lead, through several intermediary steps, to the overproduction of WbpA and WbpP, resulting in better attachment to graphite anodes and thus higher current densities.
|
|
Scooped by
?
June 10, 3:52 PM
|
The fungal kingdom is a diverse eukaryotic clade largely composed of microbial organisms. Comprised of indispensable plant symbionts, major environmental decomposers, and important plant and animal pathogens, fungi directly shape the biological world. Advancements in genomics have increased our understanding of fungal genomic variation, the mechanisms that generate it, and the association of genomic plasticity with fungal ecological lifestyles and adaptation. In this review, we discuss the patterns and mechanisms underlying fungal genomic plasticity, juxtaposing them, where known, with patterns and mechanisms observed in plants and animals. We begin by describing the plasticity of fungal genomes, which ranges from large-scale variation in size and ploidy to the physical or temporal compartmentalization of evolutionary rates. We summarize notable patterns of genomic plasticity that have evolved independently across the fungal kingdom, including but not limited to accessory chromosomes, two-speed genomes, and hypermutation; some of these patterns are unique to fungi and largely absent from animals or plants. Strikingly, certain patterns are associated with specific ecological lifestyles, raising the hypothesis that the plasticity of fungal genomes facilitates these lifestyles. We then describe the wide variety of mechanisms that contribute to fungal genome plasticity, including sexual reproduction, transposable-element mobilization, and aneuploidy. Some mechanisms appear to be lineage-specific (for example, Starship transposable elements and repeat-induced point mutations), whereas others are more broadly distributed (parasexual and sexual reproduction). We conclude by discussing how a more balanced sampling of fungal genomes, including population genomic data, will lead to greater insights into fungal genome plasticity and its origins.
|
|
Scooped by
?
June 10, 3:36 PM
|
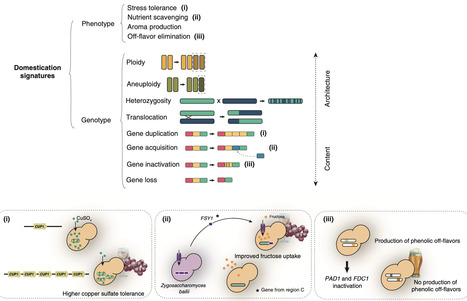
Like plants and animals, microorganisms have also been domesticated since ancient times by humans. Domesticated yeasts are required for wine, beer and bread production, among many other food and beverage fermentations. In this review, the term ‘domesticate’ refers to an organism propagated by humans that is genetically and phenotypically distinct from its wild ancestors in ways that make it more useful to humans. A single species, Saccharomyces cerevisiae, is responsible for most yeast fermentations and separate lineages within this species carry out different fermentations. Besides alterations at the phenotypic level, domestication can lead to modifications in genome architecture and genome content. These changes, which can be viewed as domestication signatures, will be briefly reviewed here. We also present and discuss special cases of domestication like ‘secondary domestication’, the situation in which an already domesticated yeast endures a second round of artificial selection, and ‘quasi-domestication’, which corresponds to evidence of adaptation of a microorganism to an environment created by humans without a detectable positive impact on the characteristics of the fermented product. Finally, we analyze other cases of confirmed or possible domestication in other Saccharomyces species and among species outside of this genus, like Brettanomyces bruxellensis and Torulaspora delbrueckii.
|
|
Scooped by
?
June 10, 3:07 PM
|
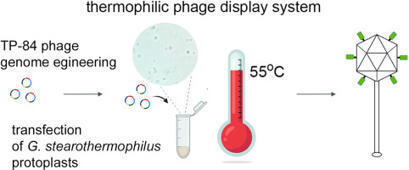
Phage display technology is a powerful technique that allows to expose any peptide fused to a bacteriophage coat protein on the surface of the virion. However, the phage display systems have limitations that impair their applications in microbiology and biotechnology. We present the construction of the first thermophilic phage display system, including ‘mosaic’ system, and provide examples of its biotechnological usefulness. The system relies on TP-84 bacteriophage, infecting Geobacillus stearothermophilus, with proliferation temperature up to 73oC, and developing only in the lytic mode, which allows liberating virion with any attached peptide. TP-84 has a large capsid, tolerates changes in the capsid proteins arrangement and develops in the thermophilic host, preventing recombinant protein aggregation in the cytoplasm. Furthermore, we introduce ‘affinity coupling’ functionalized bionanoparticle system, allowing attachment of theoretically any size proteins or even non-proteinous ligands onto TP-84 capsid, overcoming genome/capsid size limitations. The system provides technology for generation of novel types thermostable, biodegradable biomaterials. Moreover, the duplicated major capsid protein phage was constructed, forming the first thermophilic phage gene expression system. Upon a replacement of the engineered TP84_12 gene copy by a gene to be expressed, high level recombinant proteins expression can be achieved. This versatile phage display system should be useful in many microbiological approaches, overcoming the drawbacks of previous systems.
|
|
Scooped by
?
June 10, 2:50 PM
|
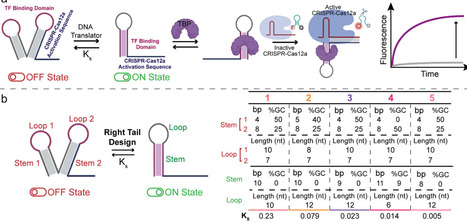
CRISPR-Cas systems have advanced many domains in life sciences, enabling diverse applications in gene editing, diagnostics, and biosensing. Here, we introduce a platform that leverages transcription factors (TFs) to regulate CRISPR-Cas12a trans-cleavage activity via engineered DNA translators. These dynamic DNA structures respond to TF binding by switching conformations, modulating Cas12a activity. Using TATA-binding protein and Myc-Max as TF models, we optimized DNA translators for precise and tunable control with rapid response kinetics. We demonstrated the platform’s specificity and versatility by integrating TF-induced regulation into synthetic biology networks, including the activation of a fluorogenic RNA aptamer (Mango III) and the creation of an artificial multimolecular communication pathway between Cas12a and Cas13a. This work establishes TFs as effective regulators of CRISPR-Cas systems, enabling novel protein-nucleic acid communication channels, showing potential for novel synthetic biology applications.
|
|
Scooped by
?
June 10, 2:19 PM
|
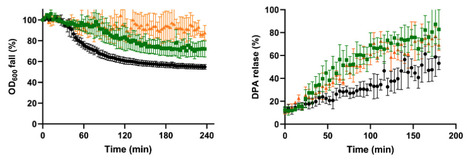
While the effect of sporulation temperature on spore germination has been extensively studied, the effect of other relevant environmental factors such as water activity (aw) has been overlooked, despite the fact that sporulation niches with lower humidity than ideal culture media, such as soil, are common in nature. In this work, we characterized the germination kinetics of B. subtilis 168 spores produced at reduced aw (0.98) using either glycerol (Sgly spores) or NaCl (Ssalt spores) in comparison to spores prepared under optimal conditions (Scontrol spores aw ~0.99) in a variety of nutrient and chemical stimuli, along with the effect of thermal activation. Spores produced at reduced aw showed impaired germination to varying degrees depending on the nutrient and solute used to depress aw. While Sgly spores exhibited germination defects in all the nutrients tested (a rich growth medium, L-alanine, L-valine, and the AGFK mixture) compared to Scontrol spores, Ssalt populations showed an impaired response to L-alanine and L-valine. These nutrient germination defects of the Sgly and Ssalt spores could not be reversed by heat activation. In addition, both populations displayed impaired germination in Ca-DPA, but increased germination rate in dodecylamine. The phenotypes of spores produced at reduced aw suggested plausible alterations in their coat properties. Using mutant spores with coat morphogenesis defects and a 4 kDa FITC-dextran probe permeability test, we could infer that the impaired germination of Ssalt spores in nutrients and Ca-DPA may involve alterations in the crust and/or outer coat, leading to reduced permeability.
|
|
|
Scooped by
?
Today, 1:25 AM
|
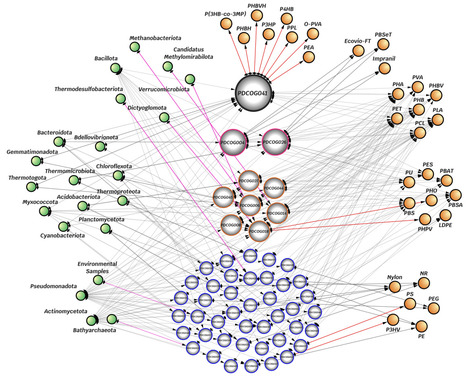
Microplastics and nanoplastics pollution (MNPP) is a major environmental issue due to its small size, long-lasting nature, and potentially harmful impacts on ecosystems and human health. Plastics can be degraded in nature by physical, chemical, and biological processes. Plastic biodegradation is a potential sustainable solution to MNPP, but understanding the fitness of potential plastic-degrading proteins (PPDPs) across environments requires comparative resources. Here, we introduce the Plastic-Degrading Clusters of Orthologous Groups (PDCOGs) database, available at https://phylobone.com/microworld/PDCOG The resource includes 625,616 PPDPs from free-living prokaryotes classified in 51 PDCOGs. The PDCOG database is a robust resource that improves understanding of plastic biodegradation and supports research on global MNPP challenges. Overall, PDPPs represent 3.5% of proteins in prokaryotes and are widely distributed across the Earth, spanning across most prokaryotic species and environments. Nearly all prokaryotic species (>95%) have the potential to biodegrade at least one polymer type.
|
|
Scooped by
?
Today, 1:06 AM
|
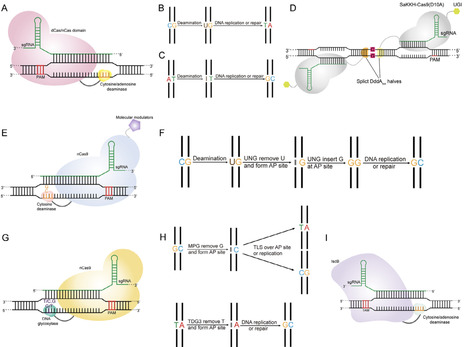
Base editing technology is a novel gene-editing approach derived from the CRISPR/Cas9 system, enabling precise and efficient base conversion. Due to operational simplicity, strong target specificity, high editing efficiency, and minimal editing byproducts, base editing has been widely applied in gene therapy, crop breeding, construction of model organisms, and microbial metabolic engineering. In this review, we systematically summarize the development history and recent advancements of several major base editors, including cytosine base editors (CBEs), adenine base editors (ABEs), CRISPR-free base editors, C-to-G base editors (CGBEs), glycosylase base editors (GBEs), and IscB-derived base editors. Furthermore, we comprehensively summarize optimization strategies for base editors and its application in constructing efficient industrial microorganism, such as Bacillus subtilis, Corynebacterium glutamicum, Saccharomyces cerevisiae, Yarrowia lipolytica, and Aspergillus niger. This review aims to facilitate the broader application of base editing technologies in synthetic biology and accelerate their translational potential.
|
|
Scooped by
?
Today, 12:57 AM
|
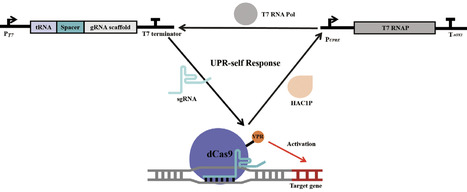
The CRISPR activation (CRISPRa) transcriptional system has become a powerful synthetic biology tool for the regulation of endogenous gene expression, allowing for precise fine-tuning of target genes through the simple modification of sgRNA sequences. In this study, we demonstrate that sgRNAs can be effectively expressed using the T7 transcription system. The insertion of tRNA sequences between PT7 and sgRNAs significantly enhances the efficiency of transcriptional activation. Furthermore, the design of PT7-tRNA-sgRNA arrays facilitates the multiplexed activation of genes. sgRNA expression was regulated by the Tet-on induction system, split-T7 system, and RNA cleavage processing by HH-HDV, resulting in the creation of a Boolean logic gene circuit capable of performing both AND and OR logic operations. Finally, we developed a UPR self-responsive system by utilizing endogenous promoters that are responsive to UPR signals to control the expression of T7 RNAP. This system dynamically regulates the expression of the endogenous HAC1 transcription factor, thus enhancing the secretion of heterologous proteins. The findings from this study highlight the potential of utilizing the T7 transcription system for the construction of genetic circuits, providing a practical toolkit for gene regulation in the industrial Komagataella phaffii strain.
|
|
Scooped by
?
Today, 12:38 AM
|
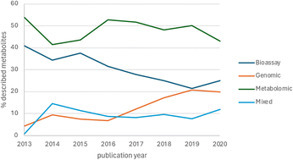
In this review, we analyzed the scientific literature of the period 2013–2023 that reported novel specialized metabolites from the Actinomycetes, one of the most prolific producers of natural products. The discovered metabolites were categorized on the basis of their chemical originality into two groups: variants of known molecules, or original metabolites. In addition, we subdivided the approaches used to discover these metabolites into four categories: bioassay-based screening, genome mining, metabolome mining, or combinations thereof. We present selected examples of the different approaches used and the resulting original metabolites. Finally, we measure the overall trends of discovery in terms of approaches, of the frequency of original metabolites and of the major biosynthetic classes that have been described. Overall, our analysis indicates that new metabolites continue to be discovered from Actinomycetes at a relatively constant rate and that the frequency of original metabolites seems to be approach-independent and relatively constant within the analyzed time period.
|
|
Scooped by
?
June 10, 11:26 PM
|
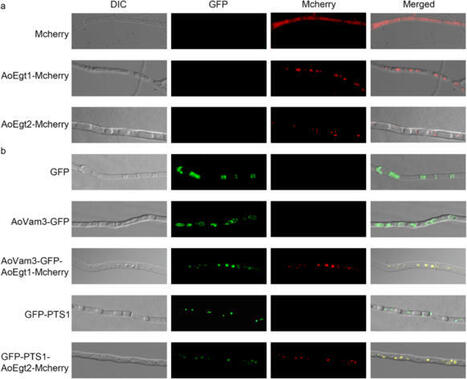
Ergothioneine (EGT) is a rare amino acid with potent antioxidant and anti-inflammatory properties, with a wide range of applications in food, cosmetics, and medicine. In the present study, Aspergillus oryzae, a common edible fungus, was engineered as an optimal host for EGT production. Moreover, two endogenous genes involved in EGT biosynthesis were characterized. The homolog AoEgt1 was shown to be localized in the vacuoles, whereas the homolog AoEgt2 was found in the peroxisomes. Overexpression of EGT biosynthetic genes from different organisms enhanced EGT production, yielding 15.17 mg EGT/g of dry weight. Using glucose as the carbon source and supplementing methionine (Met) as a precursor further increased EGT production to 20.03 mg EGT/g of dry weight, constituting an eight-fold increase compared to the wild-type strain. This study discusses the successful construction of a high-yielding A. oryzae strain for EGT biosynthesis, providing a novel strategy for efficient EGT synthesis.
|
|
Scooped by
?
June 10, 11:16 PM
|
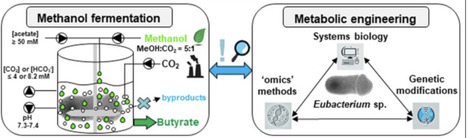
Acetogens are anaerobic bacteria of special interest in fighting environmental and economic impacts caused by massive carbon emissions that pollute our Earth’s atmosphere. These microbes have the unique ability to convert carbon monoxide, carbon dioxide, or methanol into value-added bioproducts. The gas fermentation technology is already at industrial scale and is making use of special acetogens able to produce ethanol natively. Here, we propose a methanol-based bioconversion process using anaerobic methylotrophic acetogens (Eubacterium limosum or Eubacterium callanderi) to produce natively only butyrate or butanol, if genetically modified. Therefore, we depict the crucial fermentation parameters and explain the underlying metabolic pathways to steer these biocatalysts towards sole butyrate production. Additionally, the available genetic toolkits are outlined, and the insights gained via system biology approaches are presented. The concept of the suggested bioprocess is only sustainable if green methanol is used as one-carbon feedstock. The use of black or gray methanol would undoubtedly counteract all efforts towards net-zero CO2 emissions. To meet tightening climate targets and environmental, social, and governance commitments, stakeholders must evaluate a spectrum of low‑carbon technologies. The data presented here indicate that biotechnological fermentations can reduce emissions while remaining commercially competitive, and therefore warrant serious consideration for future industrial deployment and investment.
|
|
Scooped by
?
June 10, 10:41 PM
|
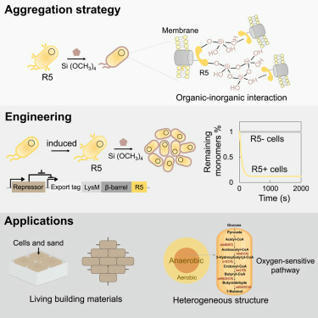
Programmable cell aggregation offers valuable insights into the natural development of synthetic multicellular systems and enables precise control over spatial organization and material structuring. Previous efforts have focused on modifying cells with designed organic-based adhesive modules and assembling cells into defined patterns. Here, we present a different approach to guide cell assembly by tuning the organic-inorganic interactions. Our method involves engineering cells to express a silicifying peptide on their surfaces, which promotes silica deposition on the cell walls. This peptide simultaneously binds to the silica synthesized on adjacent cells, triggering cell clustering. The engineered cells exhibit rapid aggregation, with approximately 95% of cells assembling within 15 min. We further show that this capability can facilitate materials assembly and chemical production. Our biosilicification-based approach offers novel insights into natural multicellularity mechanisms and holds potential for applications in biomanufacturing and materials engineering.
|
|
Scooped by
?
June 10, 4:03 PM
|
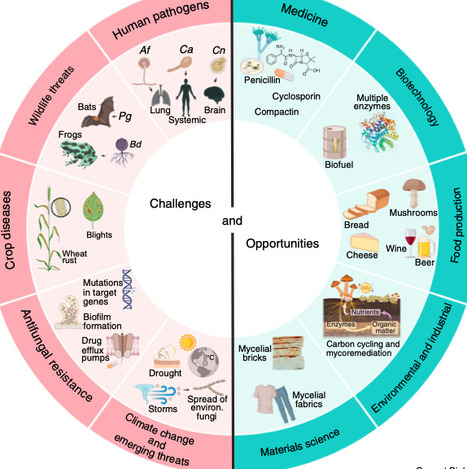
The Fungal Kingdom is vast, spanning two to five million, or even more, fungal species. Only a relatively small fraction has been isolated and named, and an even smaller number have been subjected to genetic, molecular, and genomic studies. This special issue of Current Biology celebrates the diversity of the fungal kingdom, confronts some of the serious challenges presented by fungi, and highlights the wealth of opportunities they present for biological discovery.
|
|
Scooped by
?
June 10, 3:40 PM
|
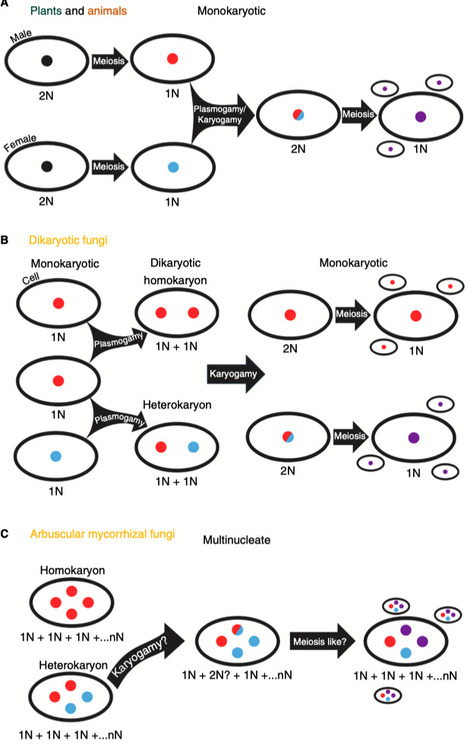
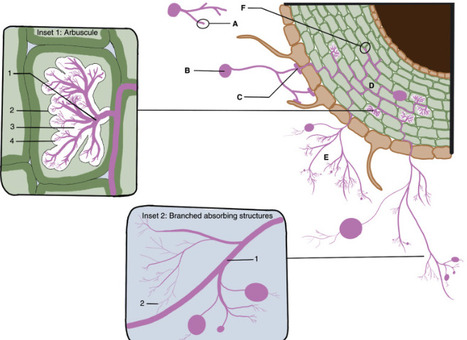
Arbuscular mycorrhizal (AM) fungi are ancient plant mutualists that are ubiquitous across terrestrial ecosystems. These fungi are unique among most eukaryotes because they form multinucleate, open-pipe mycelial networks, where nutrients, organelles, and chemical signals move bidirectionally across a continuous cytoplasm. AMF play a crucial role in ecosystem functioning by supporting plant growth, mediating ecosystem diversity, and contributing to carbon cycling. It is estimated that plant communities allocate ∼3.93 Gt CO2e to AMF every year, much of which is stored as lipids inside the fungal network. Despite their ecological significance, the cellular biology of AMF remains underexplored. Here, we synthesise the current knowledge on AMF cellular structure and organization. We examine AMF development at different biological levels — the hypha and its content, hyphal networks and AMF spores — and explore key cellular dynamics. This includes cell wall composition, cytoplasmic contents, nuclear and lipid organisation and dynamics, network architecture, and connectivity. We highlight how their unique cellular arrangement enables complex cytoplasmic flow and nutrient exchange processes across their open-pipe mycelial networks. We discuss how both established and novel techniques, including microscopy, culturing, and high-throughput image analysis, are helping to resolve previously unknown aspects of AMF biology. By comparing these insights with established knowledge in other, well-studied filamentous fungi, we identify critical knowledge gaps and propose questions for future research to further our understanding of fundamental AMF cell biology and its contributions to ecosystem health.
|
|
Scooped by
?
June 10, 3:24 PM
|
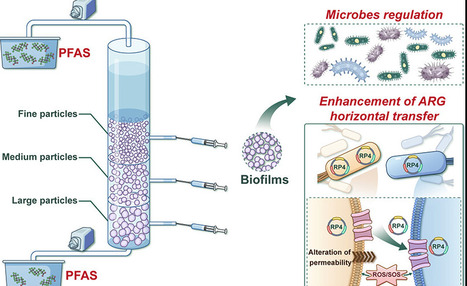
The dissemination of antibiotic resistance genes (ARGs) induced by perfluoroalkyl and polyfluoroalkyl substances (PFAS) and their ecological impacts have gained significant attention. Periphyton communities on sediments play crucial hydroecological roles and serve as bioindicators of PFAS contamination. However, research on their microbial structure and ARG dissemination in response to PFAS remains limited. This study explored how PFAS stress influences periphyton communities’ ecological functions and ARGs dynamics. PFAS varying exposure inhibited communities’ formation by decreasing biomass (3.0–26.2%) and significantly reducing protein and polysaccharides (p < 0.05) of periphyton communities. Methanogenic archaea abundance increased by 4.79–159290 times, while Variovorax and Nitrospira decreased by 1266.1–2303.5 and 36.1–140.4 times, respectively. Notably, PFAS enhanced ARGs families (multidrug, aminoglycoside, and glycopeptide) and subtypes (macB, evgS, tetA58, and bcrA), strengthening correlations between the mobile genetic elements (MGEs) and antibiotic efflux (R2 = 0.941) or target alteration (R2 = 0.961). Horizontal gene transfer (HGT) mediated by MGEs played a dominant role in ARGs dissemination compared to vertical gene transfer in periphyton communities. Mechanistic insights revealed that PFAS-induced reactive oxygen species elevation, increased membrane permeability, enhanced energy provision, and overexpression of adherent molecular genes collectively facilitated HGT-driven ARGs spread. This study provides new insights into the complex interactions between PFAS and ARGs and its potential risks in microbial habitats.
|
|
Scooped by
?
June 10, 3:00 PM
|
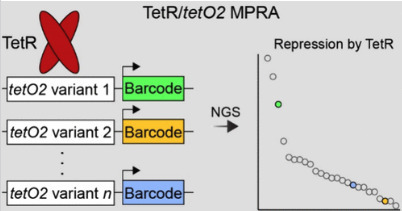
Chemogenetic regulators of transgene activity, such as the tetracycline-inducible system derived from the tetracycline resistance operon of the bacterial transposon Tn10, are critical and widely used systems in cellular engineering. The tetracycline-inducible system is prized for its selectivity, high affinity, inducibility, reversibility, and differential control of gene transcription. However, its optimization for binary on/off expression limits its application in systems biology and the modeling and construction of complex regulatory systems with intricate input/output paradigms. To overcome this limitation, we developed a high-throughput reporter system to investigate a saturated mutagenesis library of tetracycline resistance operator variants. Using this system, we mapped the functional interactions of Tet repressor DNA binding protein at single-nucleotide resolution in mammalian cells. Our comprehensive screen revealed a spectrum of variant effects, ranging from a nearly complete loss of repression to levels indistinguishable from the natural operator, validated through orthogonal assays. This comprehensive characterization of the sequence-specificity of a tetracycline resistance operator facilitates the construction of variably suppressive, inducible systems for dynamic and modular control over gene expression in mammalian cell culture.
|
|
Scooped by
?
June 10, 2:30 PM
|
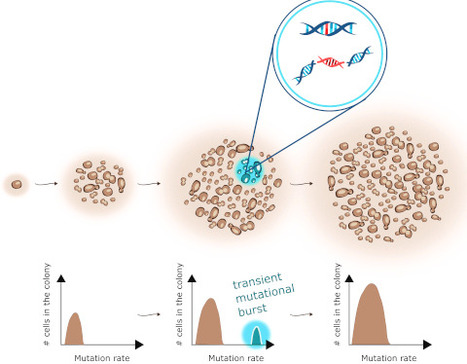
Characterizing the contribution of mutators to mutation accumulation is essential for understanding cellular adaptation and diseases like cancer. By measuring single and double mutation rates, including point mutations, segmental duplications, and reciprocal translocations, we found that wild-type yeast colonies exhibit double mutation rates up to 17 times higher than expected from experimentally determined single mutation rates. These double mutants retained wild-type mutation rates, indicating they originated from genetically normal cells that transiently expressed a mutator phenotype. Numerical simulations suggest that transient mutator subpopulations likely consist of less than a few thousand cells, and experience high-intensity mutational bursts for less than five generations. Most double mutations accumulated sequentially across cell cycles, with simultaneous acquisition being rare and likely linked to systemic genomic instability. Additionally, we explored the genetic control of transient hypermutation and found that the excess of double mutants can be modulated by replication stress and the DNA damage tolerance pathway. Our findings suggest that transient mutators play a significant role in genomic instability and contribute to the mutational load accumulating in growing isogenic populations.
|












Convergent evolution occurs when the same trait arises independently on the tree of life.