 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
Antagonistic systems of bacteria are often tightly regulated. The human gut Bacteroidales harbor three distinct antagonistic type VI secretion systems (T6SS), one of which is present only in Bacteroides fragilis, known as the GA3 T6SS. Although this is the best studied of the three T6SSs, little is known about how it is regulated. The gene upstream of the GA3 T6SS locus encodes a TetR family transcriptional regulator (TetRGA3), which we show represses expression of the GA3 T6SS locus. The gene immediately upstream and divergently transcribed from tetRGA3, designated here as lgsGA3, encodes a product of the α-oxoamine synthase family of pyridoxal phosphate-dependent enzymes with structural homology to the CqsA autoinducer synthase of the CAI-1 quorum sensing system of Vibrio spp. When lgsGA3 is deleted, transcription of the GA3 T6SS locus is repressed in a TetR-dependent manner. Strains synthesizing LgsGA3 produce a molecule released into the supernatant that likely serves as the TetRGA3 ligand, overcoming TetR transcriptional repression of the GA3 T6SS. We show that GA3 T6SS-specific immunity genes present on two acquired immunity defense islands are also regulated by LgsGA3 coordinating expression of GA3 T6SS antagonism with protection from competitor’s GA3 T6SS toxins. Production and firing of the GA3 T6SS and subsequent antagonism occurs in bacteria deleted for lgsGA3 when growing with bacteria containing this gene or their supernatants or when cocolonizing gnotobiotic mice. These data show that the GA3 T6SS is regulated by a small molecule acting through TetRGA3 allowing the bacteria to coordinate antagonistic and protective systems.
|
|
Scooped by
mhryu@live.com
Today, 12:12 AM
|
RNA-targeting CRISPR-Cas13 enzymes are robust RNA knockdown tools with both on-target and collateral cleavage activities. However, to date, the in vivo RNA cleavage mechanisms remain poorly understood. Here, we combine in vitro and in vivo methods to elucidate the exact cleavage sites of Cas13. We reveal that some subtypes of Cas13, including Cas13b and Cas13bt, cleave the target RNA at predominant positions, and rational engineering of Cas13 further improves precision. Building on these findings, we develop RNA segment editing (RSE), a targeted RNA cleavage and repair method, to restore dysfunctional RNA in cells. We anticipate that RSE will enable precision RNA engineering for therapeutics and basic research. CRISPR-Cas13 are robust RNA knockdown tools with both on-target and collateral cleavage activities. Here, the authors combine in vitro and in vivo methods to elucidate the exact cleavage sites of Cas13. The authors further develop RNA segment editing to precisely edit dysfunctional RNA in cells.
|
|
Scooped by
mhryu@live.com
April 8, 11:43 PM
|
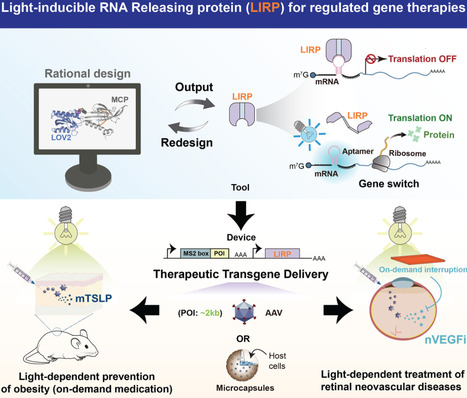
An optogenetic gene switch based on a light-inducible RNA-releasing protein (LIRP) was designed to comply with clinically licensed gene- and cell-based therapy products at a high standard. This LIRP-dependent gene switch is compatible with at least three major administration routes and/or delivery sites in vivo, such as intravitreal and intradermal expression using adeno-associated virus (AAV) vectors, as well as subcutaneous implantation of human induced pluripotent stem cell (iPSC)-derived cell grafts that stably express the corresponding genetic componentry. Using an exemplary case of iPSC-to-hepatocyte conversion, we show that the performance of the light-inducible gene switch was even improved following the differentiation process. This suggests that LIRP could permit robust and consistent regulation profiles across a wide range of cellular environments, which offers the perspective of manufacturing a variety of cell therapy products using different therapeutic chassis. In the particular case of intravitreal expression, a LIRP-dependent gene switch could provide an essential ‘safety upgrade’ for retinal gene therapies, where conventional strategies in the clinics would use AAV vectors to achieve constitutive production of vascular endothelial growth factor (VEGF) inhibitors for the treatment of wet age-related macular degeneration (e.g., NCT05407636). Here, by using the same AAV vectors to deliver a LIRP-dependent gene switch regulating intravitreal expression of similar VEGF inhibitors, we show how a light-regulated treatment regimen could achieve similar therapeutic efficacy as with constitutive expression strategies, but with markedly reduced adverse effects that may result from excessive VEGF inhibition. As LIRP was designed to be compatible with various clinically accepted gene therapy products, it may require minimal safety and efficacy testing as well as modest profitability analyses to establish a new alternative for retinal neovascular disease therapies. Thus, we believe that the overall maturity of the present technology may approach Technology Readiness Level (TRL) 5/6 levels from the viewpoints of intellectual elaboration and overall innovative impact.
|
|
Scooped by
mhryu@live.com
April 8, 11:16 PM
|
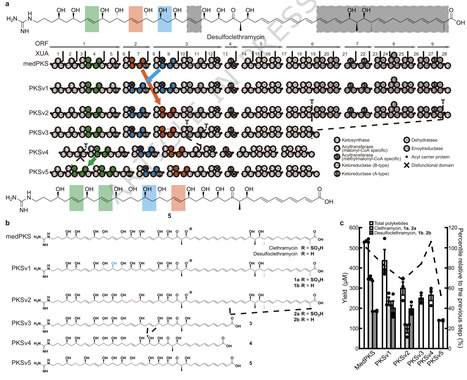
Assembly line biosynthesis creates numerous structurally diverse natural products using a common modular synthetic strategy. The collinearity between the architectures of modular polyketide synthases (PKS) and the structures of their polyketide products would seem to render these biosynthetic machineries excellent platforms for designer biosynthesis, yet reliable strategies to reprogram these assembly lines without diminishing their activities have not been identified. Here, as a best practice for PKS engineering, we demonstrate the reprogramming of the mediomycin PKS without significant loss of productivity. Using in vitro CRISPR/Cas9 gene editing followed by heterologous expression, we reconstruct an inaccessible drug lead of the fibrinogen receptor, tetrafibricin, at 82 ± 3 mg/L yield, retaining 26% productivity after five-step module editing using an evolution-supported cut site, downstream of the acyltransferase domain. A macrocyclic aminopolyol is also accessed through thioesterase swapping. These results pave the way toward the rational reprogramming of PKSs to access desired complex organic molecules. The collinearity between the architectures of modular polyketide synthases (PKS) and the structures of their polyketide products would suggest these biosynthetic machineries are excellent platforms for designer biosynthesis, yet reliable strategies to reprogram these assembly lines without diminishing their activities have not been identified. Here, the authors demonstrate the reprogramming of the mediomycin PKS without significant loss of productivity, and reconstruct an inaccessible drug lead of the fibrinogen receptor, tetrafibricin, at 82 mg/L yield.
|
|
Scooped by
mhryu@live.com
April 8, 10:44 PM
|
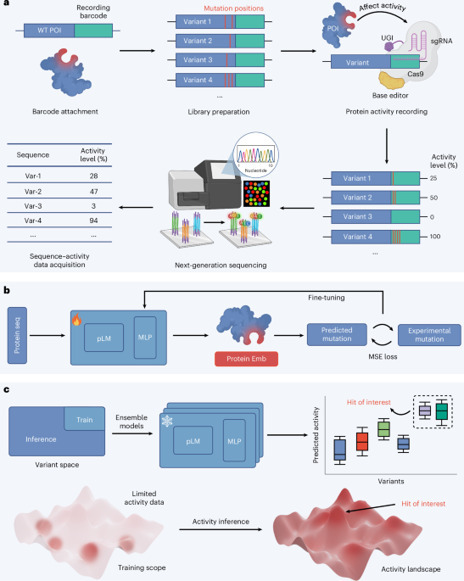
Engineering proteins with desired functions remains challenging and usually requires multiple rounds of screening and selection. Here, we present Sequence Display, a platform that generates large-scale protein sequence–activity datasets in a single round. Sequence Display enables multiplexed assessment of individual variant activity within a single experiment, offering a robust approach to mapping detailed sequence–function relationships. We demonstrate the platform’s broad applicability by generating datasets for cytosine deaminase, uracil glycosylase inhibitor, aminoacyl-tRNA synthetase and a compact Cas9 nuclease. Integrating these datasets obtained from Sequence Display with pretrained protein language models, fine-grained, variant-specific activity landscapes can be constructed. We discovered several Cas9 variants with expanded protospacer-adjacent motif recognition and evolved aminoacyl-tRNA synthetase variants capable of recognizing different noncanonical amino acids. Together, this study establishes Sequence Display as a powerful tool for mapping protein activity landscapes and accelerating the discovery of optimized proteins for biological and medical applications. Sequence Display maps protein variant activities to a sequencing-based readout.
|
|
Scooped by
mhryu@live.com
April 8, 10:30 PM
|
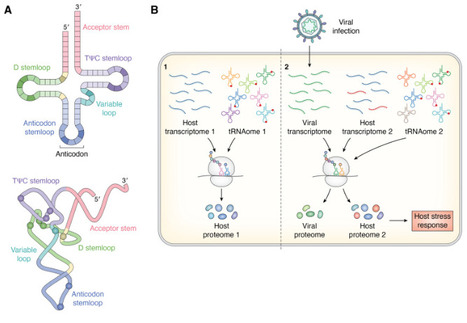
tRNA modifications are key players in post-transcriptional gene expression regulation under external and cellular stresses. Viral infection is a common external stress that hijacks cellular processes for replication. Host cell tRNA pools and modification profiles are often reshaped during viral infection, eliciting both pro- and anti-viral effects. Changes in the host tRNA modifications, particularly in the anticodon sequence, have the capability to reprogram host and viral proteomes. For example, anticodon loop modifications contribute to programmed ribosomal frameshifting essential for producing certain viral proteins. However, the roles of tRNA modifications are not limited to translation during viral infection. Retroviruses use select host cell tRNAs as reverse transcription primers and modifications modulate steps in reverse transcription. Furthermore, some non-primer modified tRNAs are selectively packaged into virion particles, though their functions remain unknown. Virally-encoded tRNAs harbor modifications that expand the anticodon pool, as host tRNAs are depleted. Expression and activity of several tRNA-modifying enzymes are regulated upon viral infection, the functional implications of which remain to be elucidated. tRNA modifications are involved in anti-viral defense, particularly in tRNA cleavage at the anticodon loop following viral infection, leading to tRNA-derived fragments. While the tRNA modification landscape is likely significantly altered following most viral infections, current evidence is limited to a few specific examples. A global tRNAome analysis during viral infection will shed light on the regulation of these and other processes. Emerging technologies including advances in direct tRNA sequencing and modification detection via mass spectrometry are making this possible.
|
|
Scooped by
mhryu@live.com
April 8, 4:49 PM
|
Type VII CRISPR-Cas system, evolutionarily associated with type III systems, utilizes a Cascade complex formed by Cas5 and catalytically inactive Cas7 copies for target RNA binding, but instead incorporates a specialized Cas14 ribonuclease for target cleavage. Here, we report a high-quality cryo-EM structure at the target engagement state with a shortened crRNA and elucidate how the recruited Cas14 captures the target RNA and undergoes target-mediated activation. The signature Cas14 is homologous to eukaryotic CPSF73 and prokaryotic RNase J, comprising two conserved subdomains, MβL and β-CASP. Different from canonical type III systems, 5′-end target RNA, rather than 3′-end, is bent into the positively charged binding channel formed by the two subdomains to access the conserved catalytic pocket on Cas14. Two special structural features, α1 helix from Cas7 and α10 helix from Cas14, promote the bent target RNA docking into the catalytic pocket of Cas14 nuclease in concert. A dual-functional loop, displaced by the entering target RNA, induces a closed-to-open transition between the two subdomains for nuclease activation. More importantly, the flipped dual-functional loop also maintains the stabilization of incoming target RNA. Altogether, our work provides a more comprehensive understanding of type VII system mechanism, laying a mechanistic foundation for RNA-targeting tool development.
|
|
Scooped by
mhryu@live.com
April 8, 4:32 PM
|
Bacterial exposure to constant phage attack drives rapid diversification of antiphage defense systems, often through the exchange of modular defensive domains. Here, we leverage this modularity signature to identify new defense systems by systematically searching for operons encoding known defensive domains in non-canonical configurations. Using this approach, we identified 39 848 candidate defense operons in E. coli genomes. Annotation of the operons based on their shared defensive domains with known systems reveals that the operons represent variants of 82 defense families. Experimental testing of nine candidates validated six with anti-phage activity. These include DarTG and ietAS system variants that have acquired helicase modules, and a Gabija system in which a MazF-like protein replaces GajA, implying novel anti-phage mechanisms. We also identified a new clade of Pycsar that synergizes with type IV Thoeris to broaden phage protection. Our findings demonstrate that mining modular defensive domains provides a powerful strategy to predict and characterize new anti-phage systems, expanding the known repertoire of bacterial immunity.
|
|
Scooped by
mhryu@live.com
April 8, 2:52 PM
|
Protein functional annotation is essential for understanding biological processes, disease mechanisms, and enzyme activities, yet experimental validation remains costly and low-throughput. With the rapid development of Artificial Intelligence (AI), a wide range of computational approaches have been proposed to infer protein functions. This review systematically examines methods for annotating Gene Ontology (GO) terms and Enzyme Commission (EC) numbers. These are two complementary systems that capture different aspects of protein functions. Based on these two systems, we first synthesize existing approaches into six general modeling paradigms with a clear, structured framework. Then, we introduce GO and EC in a parallel manner, consisting of representative methods, commonly used evaluation metrics, prediction scenarios, and task-specific challenges. Finally, we outline emerging opportunities and future directions aimed at achieving more accurate, context-dependent, and high-resolution protein functional annotation.
|
|
Scooped by
mhryu@live.com
April 8, 2:48 PM
|
Quorum sensing enables bacteria to coordinate gene expression in response to population density through the detection of small-molecule signals. In gram-negative bacteria, LuxR-type transcription factors bind acyl-homoserine lactones to regulate collective behaviors, yet how ligand sensitivity is tuned to shape transcriptional outcomes remains poorly defined. In Pseudomonas aeruginosa, the quorum-sensing receptor RhlR responds to N-butyryl-L-homoserine lactone (C4HSL) and controls late-stage quorum-sensing behaviors, including phenazine biosynthesis. Here, we use a chemical-genetic and structure-guided mutational approach to define how RhlR ligand sensitivity regulates promoter-specific transcription. We identify substitutions within the RhlR ligand-binding pocket that enhance sensitivity to C4HSL without altering ligand specificity, generating hypersensitive receptor variants. Increased ligand sensitivity selectively represses phenazine biosynthetic gene expression, reduces pyocyanin production, and alters phenazine output, while leaving other RhlR-dependent quorum-sensing traits unaffected. Transcriptomic and chromatin immunoprecipitation analyses reveal that these effects arise from reduced expression of the RhlR co-regulator PqsE, leading to decreased RhlR occupancy at phenazine gene promoters. These findings support a coincidence-detection mechanism in which ligand sensing and co-regulator availability jointly determine transcriptional output. Together, our results demonstrate that ligand sensitivity is a critical regulatory determinant that tunes quorum-sensing gene expression. This work reveals how changes in signal detection can reshape transcriptional hierarchies and metabolic outputs, providing insight into the fine control of bacterial collective behaviors and virulence-associated metabolism.
|
|
Scooped by
mhryu@live.com
April 8, 2:36 PM
|
The 5′-repeat fragments released during pre-crRNA maturation are critical yet understudied components of the CRISPR/Cas12a system. Here, we demonstrate that engineered 5′-repeat fragments can potently activate Cas12a cleavage, with efficiency strongly dependent on the length of the 3′-spacer. Strikingly, complete truncation of the 3′-spacer generates a “chiral-like crRNA” conformation that induces a delayed-switch mode of Cas12a activation, fundamentally distinct from conventional mature crRNA. Leveraging this characteristic, we develop the delayed cleavage feature-mediated one-pot sensing strategy that resolves the long-standing challenge of incompatibility between Cas12a-based cleavage reaction and nucleic acid amplification, achieving a 1000-fold improvement in sensitivity over that of the conventional mature crRNA-mediated one-pot method. Furthermore, we integrate a cleavage-based one-pot assay with a portable temperature-controlled fluorescence imaging device to create an on-site diagnostic platform for high-throughput screening. Our study further advances the understanding of the crRNA-guided mechanism and facilitates the expansion of its applications in genome editing and molecular diagnostics. In CRISPR/Cas12a systems, small RNA from pre-crRNA maturation is often overlooked. Here, authors show these fragments reconstitute with Cas12a to activate cleavage. They discover a crRNA activating Cas12a in a “delayed-switch” mode, resolving its incompatibility with nucleic acid amplification.
|
|
Scooped by
mhryu@live.com
April 8, 1:25 PM
|
Protein-protein interactions (PPI) are molecular lego which define the physical states of cells. Accurately identifying PPIs remains challenging due to the interplay of several factors ranging from electrostatic to molecular geometry, topology, and physics. Existing computational approaches capture only fragments of this orchestra, limiting their generalizability across protein families and interaction types. Here, we present ProMaya, a hierarchical multi-scale Graph-transformer framework that integrates 3D atomic geometry, electronic distribution, residue-level structure and disorder, surface mass-density signatures, and large protein language-model embeddings of interacting proteins. Highly comprehensively benchmarked across nine species and 47 GB experimentally validated data, ProMaya achieved consistently >95% average accuracy, outperforming state-of-the-art tools by >12%. As driven by its explainability, the first time introduced atomic and protein language information dramatically boosted it to an outstanding level for PPI discovery in any species, potent to even bypass costly experiments. ProMaya system is freely accessible at https://scbb.ihbt.res.in/ProMaya/
|
|
Scooped by
mhryu@live.com
April 8, 2:00 AM
|
Translational fusion of two separate genes into a compound sequence encoding a fusion protein is a key evolutionary mechanism which underpins the emergence of new protein activities, families, and architectures. In biotechnology, gene fusion is a valuable molecular evolution tool for tagging proteins of interest and for combining or altering protein activities. To broadly demonstrate and harness the gain-of-function capabilities of fusion genes in a whole-genome approach, we constructed a “Function Generator™” fusion gene library containing pairwise combinations of 5,019 protein-coding sequences in the Saccharomyces cerevisiae genome. The open reading frames (ORFs) were PCR amplified from the S288C yeast genome and cloned into a centromeric expression vector, with each ORF represented in the 5′ and the 3′ positions of the resulting gene fusions. To illustrate the ability of fusion genes in the library to confer complex phenotypes, a population of yeast library transformants was screened for resistance to four toxic heavy metal ions (Cd+2, Co+2, Cu+2, and Ni+2). Active fusion genes were cloned, validated, and sequenced, revealing a multitude of biological functions represented in these genes, including proteins involved in transcription, translation, metal ion binding and transport, and cell cycle control, as well as unknown functions. The gain-of-function principle of gene fusions was confirmed by comparing the activity of selected fusion genes to their constituent single ORFs expressed either individually or in nonfused pairs. Function Generator™ represents a powerful way to approach phenotypic diversity in the laboratory and to bypass a key evolutionary bottleneck for accelerated strain development.
|
|
|
Scooped by
mhryu@live.com
Today, 12:23 AM
|
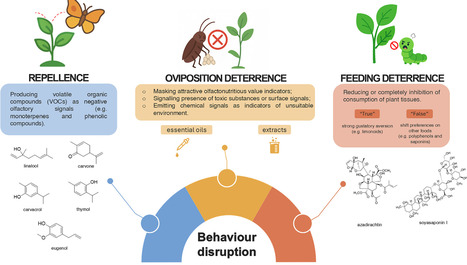
Plant secondary (specialized) metabolites play a pivotal role in disrupting pest behavior, offering a promising and environmentally friendly alternative to conventional pesticides. These compounds can interfere with insect feeding, oviposition, and host selection, thereby reducing crop damage and pest populations. Recent advances highlight the ecological selectivity and rapid biodegradation of these metabolites, making them attractive for sustainable crop protection. Innovative formulation techniques are enhancing their persistence and efficacy, yet challenges remain in understanding synergistic effects, nontarget impacts, and practical implementation. Harnessing the full potential of plant metabolites for pest behavioural disruption requires integrated research and development, paving the way for their broader adoption in integrated pest management strategies.
|
|
Scooped by
mhryu@live.com
April 8, 11:55 PM
|
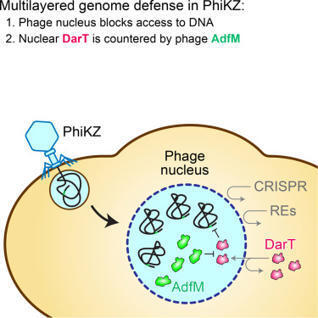
Chimallivirus bacteriophages enclose their replicating genomes in a protein-based compartment termed the phage nucleus. While the phage nucleus segregates phage DNA from host immune proteins, it is not known if additional factors are required to protect against DNA-targeting host defenses. Here, we identify a chimallivirus-encoded DarG2-like antitoxin that localizes to the phage nucleus and provides protection against phage-targeting DarTG2 toxin-antitoxin systems. This protein, which we term AdfM (anti-darT factor macro), contains a macrodomain and removes DarT2-mediated ADP-ribose modifications from DNA. In the absence of AdfM, DarT2 modifies phage DNA and restricts chimallivirus replication despite being largely excluded from the phage nucleus. Increasing the nuclear concentration of DarT2 while decreasing the nuclear concentration of AdfM reduces phage replication. These results show that the phage nucleus is insufficient to completely protect the chimallivirus genome from host defenses; rather, it is one component of a multilayered counter-defense strategy.
|
|
Scooped by
mhryu@live.com
April 8, 11:40 PM
|
Cyclic-di-GMP (c-di-GMP) is a ubiquitous second messenger in bacteria and regulates a variety of cell activities. Many bacteria contain multiple enzymes involved in c-di-GMP synthesis or degradation; however, how they coordinate with each other to orchestrate c-di-GMP homeostasis remains unclear. Here, using the cyanobacterium Anabaena PCC 7120 as a model, we created cdG0 and cdGmax strains by deleting all 8 and 14 genes, respectively, that encode enzymes with c-di-GMP degradation and synthesis domains, alongside a collection of mutants with various numbers of these genes deleted. Our findings demonstrate that c-di-GMP in Anabaena not only modulates cell size but is also indispensable for cell viability. Quantitative analysis established two critical physiological thresholds in vivo: a minimal c-di-GMP level required for cell size maintenance and a lower, lethal threshold essential for survival. We show that the 16 enzymes involved in c-di-GMP turnover in Anabaena function as an electromechanical-like dual relay to control c-di-GMP dynamics, with different modules contributing to c-di-GMP homeostasis or responding as an SOS alarm when the c-di-GMP concentration drops below the lethal threshold. Both effects of c-di-GMP on cell size reduction and cell viability are mediated by the cyclic-di-GMP receptor (CdgR), depending on the amount of the c-di-GMP-free form of CdgR available because of titration by c-di-GMP in the cells. The system, with the two concentration thresholds of c-di-GMP that dictate cell size and viability, respectively, enables dynamic cellular adaptation while preventing lethal effects.
|
|
Scooped by
mhryu@live.com
April 8, 10:52 PM
|
The 3D architecture and dynamics of the genome are crucial for regulation of genome stability, transcription and cellular function. CRISPR-based live imaging technologies have enabled real-time visualization of specific genomic loci and transcripts in living cells. These tools harness customized guide RNAs and nuclease-deactivated Cas effectors to achieve precise genomic targeting, and recent methodological advances provide the 3D spatiotemporal resolution required to decipher real-time chromatin communication. These methods are elucidating the biophysical properties of chromatin, linking dynamic enhancer–promoter interactions directly to transcription, and revealing the role of 3D genome dynamics in basic cellular processes and disease. Here, we summarize the development of CRISPR-based live-cell imaging techniques, highlight the complementary 3D microscopy and analysis methods compatible with these methods, and offer perspectives on their applications to uncover fundamental principles that govern genome dynamics and function. In this Review, Zhu et al. outline the various CRISPR-based tools recently built for dynamic DNA and RNA imaging, which have provided insights into various molecular mechanisms. The authors also discuss the important parameters to consider when using these tools, and how to approach the quantitative image analysis.
|
|
Scooped by
mhryu@live.com
April 8, 10:33 PM
|
Similar to proteins, many RNAs fold into three-dimensional (3D) structures to perform biological functions. Here we present the trRosettaRNA server, a web-based platform for automated RNA 3D structure prediction using deep learning. The primary input is the nucleotide sequence of a target RNA, with the option to upload custom multiple sequence alignments and secondary structures. The server uses an end-to-end neural network for automated 3D structure prediction, followed by an energy optimization step to resolve structural violations. As an automated server, trRosettaRNA is distinguished by its state-of-the-art modeling accuracy, flexible input options and comprehensive visualization of prediction results. trRosettaRNA has been successfully applied in various contexts, including predicting structures for Rfam families lacking known 3D structures, where representative cases of high-confidence structure predictions were found to align well with subsequent experimental observations. Utilizing up to 5 central processing unit (CPU) cores in parallel on our computer cluster, the server takes a median time of about 1 h to predict structures for RNA sequences with about 200 nucleotides. The standalone package for trRosettaRNA offers distinct advantages such as enhanced data privacy for sensitive sequences, the ability to bypass server queues and integration into high-throughput automated pipelines. Importantly, the open-source nature of the package empowers researchers to directly modify the codebase for specialized research needs or to develop derivative tools by fine-tuning the underlying neural network. The web server and standalone package of trRosettaRNA are available at https://yanglab.qd.sdu.edu.cn/trRosettaRNA/ and https://github.com/YangLab-SDU/trRosettaRNA2 , respectively. The trRosettaRNA server is a web-based platform for automated and accurate RNA 3D structure prediction using deep learning. This protocol also describes how to use the standalone package locally, which is beneficial for large-scale applications.
|
|
Scooped by
mhryu@live.com
April 8, 4:54 PM
|
Functional metagenomics has emerged as an effective tool for discovering novel enzymes directly from environmental samples, overcoming the limitations of traditional culture-based methods. In this study, we used a functional metagenomic approach on stool samples from Axis kuhlii, an endemic deer species from Indonesia, to identify active cellulases. We created an efficient workflow for expression of metagenomic sequences directly in Komagatella phaffii by combining metagenomic sequencing to investigate enzyme diversity, multiplex PCR to build a genes library, and rolling circle amplification (RCA) to streamline the cloning process, eliminating the need for intermediate Escherichia coli transformation and propagation steps. Furthermore, a semi-high-throughput screening method was used to evaluate multiple samples at once, allowing for the rapid identification of active enzymes. Using this approach, we discovered five endoglucanases and three β-glucosidases with confirmed enzyme activity. This study shows that functional metagenomics can bridge the gap between computational predictions and experimental validation, providing a reliable platform for enzyme discovery and characterization from complex environmental microbiomes.
|
|
Scooped by
mhryu@live.com
April 8, 4:43 PM
|
Transcription factors (TFs) efficiently locate their target DNA sequences by combining three-dimensional diffusion and one-dimensional sliding on nonspecific DNA. To balance rapid sliding with strong specific binding, TFs were proposed to switch between search and recognition conformations. For E. coli lac repressor (LacI), the folding of the hinge helices has been implicated in the conformational switch. Here, we tested how mutations in the hinge region impact the search speed and binding stability. Based on molecular dynamics simulations, we selected two LacI mutants favoring either search or recognition conformation. We measured the binding kinetics of the mutants both in vitro on DNA microarrays with 2479 different Lac operators and in vivo via single-molecule experiments. We identified a mutation that enhances the specificity but reduces binding strength globally, and another mutation that makes the operator binding stronger but also reduces the specificity. However, the altered specificity impacts the search time less than expected. Instead, the major effect was impaired dissociation in response to Isopropyl β-D-1-thiogalactopyranoside (IPTG) induction for the strongly binding mutant. Together with earlier reports of affinity–inducibility trade-offs in LacI, our data support the model in which the trade-off is between binding stability and inducibility rather than between speed and binding stability.
|
|
Scooped by
mhryu@live.com
April 8, 4:08 PM
|
The CRISPR-Cas12a system offers a promising platform for simple and sensitive nucleic acid diagnostics, including tumor-associated variant detection and infectious agent identification. However, its intrinsic mismatch tolerance limits its ability to accurately detect single-nucleotide variants (SNVs). Here, we introduce Structure-Disruption-Sensitive CRISPR (SDS-CRISPR), a programmable CRISPR-Cas12a approach that achieves highly precise allele discrimination. Guided by AlphaFold3 modeling and bioinformatic analysis, we uncover how split structural design and ionic modulation reconfigure Cas12a conformations, elucidating the structural basis of SNV discrimination in SDS-CRISPR. We apply SDS-CRISPR to detect IDH1WT and IDH1R132H alleles with attomole sensitivity and 0.01% variant frequency. To facilitate intraoperative use, we combine SDS-CRISPR with a lateral-flow strip and an artificial intelligence-assisted smartphone reader, enabling on-site detection within 20 min. Clinical validation with 43 glioma tissue samples shows high concordance with immunohistochemistry, while plasma cfDNA testing demonstrates mutation fractions consistent with next-generation sequencing. Beyond glioma, SDS-CRISPR generalizes across molecular targets, discriminating microRNA isoforms and identifying HIV-1 drug-resistance mutations. Together, these results establish SDS-CRISPR as a universal, mechanistically informed, and clinically actionable framework for precision molecular diagnostics.
|
|
Scooped by
mhryu@live.com
April 8, 2:51 PM
|
Despite their vital physiological roles, oxidative imbalance caused by reactive oxygen, nitrogen, sulphur, and chlorine species damages essential body macromolecules such as proteins, lipids, and nucleic acids through oxidative stress. This stress is strongly associated with cancer, inflammation, neurological and cardiovascular disorders, and other chronic human diseases. Therefore, antioxidants, natural or synthetic, that counteract oxidative damage are important, with increasing interest in their use within the pharmaceutical, food, and cosmetic industries. However, due to toxicity concerns with the synthetic variants, natural antioxidants are increasingly preferred. Extremophile-derived antioxidants, such as superoxide dismutases, catalases, peroxidases, carotenoids, and melanin, are of renewed interest due to their remarkable stability, robustness, and potency under extreme conditions of temperature, pH, and salinity. These make them better than many mesophile-derived antioxidants and excellent candidates for cost-effective biotechnological, research, and industrial processes that require high operational efficiency. This review summarises key classes of selected enzymatic and pigment antioxidants, their mechanisms of action, and their industrial relevance, with a focus on extremophilic microalgae, bacteria, and fungi. The benefits of extremophilic antioxidants are discussed alongside their current applications and existing challenges, including the need to develop efficient delivery systems, scalability issues, and limited characterisation.
|
|
Scooped by
mhryu@live.com
April 8, 2:41 PM
|
Cell-free systems have great potential for nucleic acid assays, yet universal onsite diagnostics without preamplification are limited. Here, a universal cell-free diagnostic platform termed TRACKer (Target-Responsive non-preAmplification Cell-free diagnostic Kit) has been developed for detecting multiple respiratory viral RNAs in laboratory and field settings. TRACKer integrates three modules: ribozyme allostery module, riboregulator activation module, and output module. The ribozyme allostery module enables universal target recognition through strand displacement-mediated ribozyme conformational switching. The riboregulator activation module achieves preamplification-free detection via cascade expression of reporter proteins. The output module provides swappable reporter templates for luminescent quantification and lateral flow visualization in diverse scenarios. TRACKer is a robust system that enables rapid ( < 70 min) nucleic acid detection without preamplification and demonstrates attomolar sensitivity (1-10 aM) for six respiratory viruses. Overall, TRACKer presents a promising approach to nucleic acid detection, offering a low-cost and scalable solution with potential applications in point-of-care diagnostics and beyond. Cell-free systems hold potential for nucleic acid assays but are limited by the need for pre-amplification. Here the authors develop a cell-free diagnostic platform that detects viral RNA without pre-amplification demonstrating uses on six respiratory viruses in under 70 minutes with attomolar sensitivity.
|
|
Scooped by
mhryu@live.com
April 8, 2:30 PM
|
Cell-type-specific promoters are used in gene therapy to restrict expression of the therapeutic payload. However, these promoters often have suboptimal strength, selectivity and size. Here, leveraging recent insights into the function of enhancers, we developed synthetic super-enhancers (SSEs) by assembling functionally validated enhancer fragments into multipart arrays. Focusing on the core SOX2-driven and SOX9-driven transcriptional regulatory network in glioblastoma stem cells (GSCs)1, we engineered SSEs with robust activity and high selectivity. Single-cell profiling, biochemical analyses and genome-binding data indicated that SSEs integrate neurodevelopmental and signalling-state transcription factors to trigger the formation of large multimeric complexes of transcription factors. Moreover, GSC-selective expression of a combination of cytotoxic (HSV-TK and ganciclovir) and immunomodulatory (IL-12) payloads, delivered using adeno-associated virus vectors, as a single treatment led to curative outcomes in a mouse model of aggressive glioblastoma. Notably, IL-12 induced an immunological memory that prevented tumor recurrence. The activity and selectivity of the adeno-associated virus and SSE were validated using primary human glioblastoma tissue and normal cortex samples. In summary, SSEs harness the unique core transcriptional programs that define the GSC phenotype and enable precision immune activation. This approach may have broader applications in other contexts when precise control of transgene expression in specific cell states is necessary. Synthetic super-enhancers enable specific delivery of anticancer payloads, achieving tumour elimination after a single dose in a mouse model of aggressive glioblastoma.
|
|
Scooped by
mhryu@live.com
April 8, 1:22 PM
|
Acinetobacter baumannii natural isolates encode multiple copies of E. coli DNA polymerase V (pol VEc) umuDC homologs, some with as many as four nonidentical umuC and three nonidentical umuD genes encoding twelve possible pol VAb variants. Here, we show that six of the twelve pol VAb drive spontaneous and methyl methanesulfonate-induced mutations when expressed in E. coli Mutagenesis depends on co-expression with RecAAb. Five mutagenically active pol VAb combinations assemble in vitro as stable mutasome complexes that synthesize DNA, pol VAb Mut = UmuD′2CAb-RecAAb-ATP/ATPγS. One of the mutasomes requires an A. baumannii-encoded β/τ processivity clamp for activity in vitro. Translesion DNA synthesis (TLS) occurs at T^T cyclobutane dimers, with different variants exhibiting different nucleotide misincorporation specificities. As observed for E. coli pol V and R391 ICE-encoded Rum pol, a single amino acid substitution in RecA, RecAAb M196D, abolishes A. baumannii pol V-induced mutagenesis.
|






























1str, fungal highway