 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:30 PM
|
Cofactor engineering is revolutionizing green biomanufacturing by overcoming the fundamental bottleneck between the limited supply/regeneration of cellular energy cofactors [e.g., NAD(P)H, ATP] and the high demands of efficient bioproduction. This review highlights advanced strategies, such as orthogonal systems that separate product synthesis pathways from basal metabolism, external energy sources (e.g., light or electricity) for cofactor regeneration, and material-enabled immobilization for scalable processes. These approaches enable high-yield production of diverse compounds, from specialized optically pure pharmaceuticals to bulk chemicals, by addressing critical limitations in yield, purity, and industrial scalability beyond conventional fermentation. Finally, we discuss challenges in process stability and economic viability, underscoring cofactor engineering’s potential as a versatile strategy for sustainable, next-generation biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 5:06 PM
|
We introduce mRNA-GPT, a generative model for end-to-end full-length mRNA sequence design and optimization. Unlike existing approaches that optimize isolated regions, mRNA-GPT jointly optimizes across all three regions (5' UTR, CDS, and 3' UTR) to capture long-range sequence dependencies and cross-region regulatory interactions critical for therapeutic efficacy. The model is pre-trained on 30 million full-length natural mRNA sequences across diverse species and organisms, establishing a robust foundation for sequence generation. We employ Reinforcement Learning (RL), specifically Proximal Policy Optimization (PPO) with oracle-based reward signals, to directly and iteratively optimize target properties, such as half-life and translation efficiency. mRNA-GPT supports flexible generation modes: single regions (UTR or CDS alone), full-length sequences, or generation of any region conditioned on any other region. Through multi-objective optimization, mRNA-GPT achieves Pareto-optimal designs that balance competing properties without sacrificing performance on either objective. mRNA-GPT demonstrates superior design capabilities compared to state-of-the-art methods, achieving enhanced performance in 3' UTR stability optimization, CDS translation rate enhancement, and comprehensive full-length sequence design.
|
|
Scooped by
mhryu@live.com
Today, 4:45 PM
|
Liquid-liquid phase separation (LLPS) underlies the formation of biomolecular liquid condensates (also referred to membraneless organelles, MLOs), which are essential for spatially organizing various biochemical processes within cells.Proteins that play a key role in driving condensates formation are termed phase-separating proteins (PSPs). Given experimental identification of PSPs remains labor-intensive and time-consuming, multiple computational tools have been developed based on empirical features or deep learning. In this study, we propose SSPSPredictor, a novel multimodal predictive model for PSPs with folded or intrinsically disordered structures, leveraging the fusion of sequence information from a protein language model ESM-2 and structural insights from a graph neural network GVP. Compared with existing tools, SSPSPredictor achieves balanced performance in identifying endogenous PSPs, predicting relative LLPS propensities, and recognizing key regions that drive LLPS. Moreover, SSPSPredictor exhibits good interpretability in identifying driving regions along protein sequences, although no relevant supervision was provided during training. Further predictive analysis of the human proteome using SSPSPredictor reveals that the proportion of intrinsically disordered proteins (IDPs) undergoing LLPS is significantly higher than that of folded proteins. In addition, pathogenic variants, especially those located in disordered regions, exhibit higher LLPS propensity than other mutations, uncovering a link between LLPS and diseases at the amino acid level.
|
|
Scooped by
mhryu@live.com
Today, 4:34 PM
|
APOBEC3A catalyzes cytosine-to-uracil deamination in single-stranded DNA and RNA. Physiologically, APOBEC3A functions in innate immunity and aberrant deamination is associated with cytosine mutations in enzymatically preferred YTCW substrate motifs in multiple cancers. Much less is known about the potential contribution of APOBEC3A-catalyzed RNA editing to virus and cancer evolution. Here, we present HAMMER (hairpin-based APOBEC3A-mediated messenger RNA editing reporter), a rapid luminescence-based cellular assay for measuring RNA editing by APOBEC3A. HAMMER reports APOBEC3A activity as a reduction in the ratio of firefly to renilla luciferase activity. Briefly, tandem renilla and firefly luciferase open reading frames are separated by an optimal APOBEC3A hairpin substrate, in which C-to-U editing of a CGA motif yields a UGA stop codon thus preventing translation of the downstream firefly luciferase reporter, without impacting the upstream renilla reporter. HAMMER activation is dose-responsive, catalytic activity-dependent, and specific to human APOBEC3A. A panel of herpesviral ribonucleotide reductase constructs was used to show that direct inhibition of APOBEC3A results in a dose-responsive recovery of firefly luciferase expression. HAMMER is therefore a scalable and easy-to-use method for quantifying cellular APOBEC3A RNA editing activity and characterizing inhibitors.
|
|
Scooped by
mhryu@live.com
Today, 4:07 PM
|
Reprogrammable Adenosine Deaminase Acting on RNA (ADAR) Sensors (RADARS) control RNA translation in mammalian cells, allowing for noninvasive sensing or perturbation of specific cell types based on transcriptional signatures. Upon base-pairing between a target RNA and a sensor RNA, RADARS leverages ADAR to edit a premature stop codon upstream of a gene of interest, thereby releasing translation of the desired cargo. These design principles enable sequence programmability, allowing RADARS to adapt more easily to new contexts than existing tools for targeting cell types. We describe a detailed protocol for performing experiments with RADARS, including designing, cloning and validating RADARS constructs targeting a transcript of interest. RADARS guide sequences can be designed with an intuitive web interface and cloned into existing constructs for downstream applications including imaging, sorting and sequencing. We outline recommendations for cargo choice, sensor design and ADAR system selection, enabling users to choose the best workflow depending on the desired application. Beginning with sensor design, the selection of top-performing RADARS guides can be completed in ~2 weeks, followed by a desired use case. Convenient engineering and application of RADARS for various applications enable the design and execution of various cell-targeting experiments. This protocol uses Reprogrammable Adenosine Deaminase Acting on RNA (ADAR) Sensors (RADARS) to robustly sense RNA transcripts inside eukaryotic cells, enabling detection of changes in gene expression or targeting and perturbation of specific mammalian cell types and states.
|
|
Scooped by
mhryu@live.com
April 1, 11:52 PM
|
Dynamic changes in lipid membrane composition are a common response to stress, often involving shifts in key lipid molecules. Phosphatidic acid (PA), a central precursor in lipid biosynthesis, accumulates when anionic phospholipid synthesis is blocked—lipids that are typically primary targets of membrane-active antimicrobial peptides (AMP). This raises the question of how cationic AMPs adapt to such lipid remodeling, which is especially relevant given their promise as novel therapeutics against escalating antimicrobial resistance. Their killing mechanism is often unclear. To identify ongoing processes clearly linked to bacterial cell death, six assays targeting membrane integrity and cell viability were performed alongside bactericidal measurements. These assays were conducted on E. coli and a mutant depleted of anionic phospholipids, treated with the cationic peptides melittin and LL-37. Correlation of assays generated characteristic antimicrobial profiles, providing insight into the peptides’ mechanisms. LL-37 acted independently of membrane composition, while melittin showed increased activity in the absence of anionic phospholipids. This study confirmed specific interactions with PA, but their action suggests targets beyond the membrane, as bacteria remained viable during membrane disruption but failed to form colonies. Overall, these findings indicate that both peptides can effectively handle lipid remodeling and uncover processes driving bacterial cell death.
|
|
Scooped by
mhryu@live.com
April 1, 11:37 PM
|
The functional annotation of protein sequences has undergone tremendous progress over recent years, but still too-many protein sequences remain as so-called hypothetical proteins after applying state-of-the-art genome annotation software pipelines. Here, we introduce Baktfold, a new command line software tool for the ultra-sensitive but taxon-independent fast annotation of protein sequences across the microbial tree of life. Baktfold conducts sequential protein structure-based searches against four complementary structure databases. Protein sequences are transformed into Foldseek 3Di tokens via the ProstT5 protein language model and subsequently searched against structure databases via Foldseek. All results are exported in GFF3 and INSDC-compliant flat files as well as comprehensive JSON files facilitating automated downstream analysis 100% interoperable with the popular bacterial annotation tool Bakta. We compared Baktfold's performance in terms of wallclock runtime and functional annotation of hypothetical proteins from various sources including bacterial and archaeal isolates, plasmids, metagenomic-assembled genomes and micro-eukaryotes. When benchmarked on over three hundred thousand species representatives across the prokaryotic tree of life, Baktfold;s median overall bacterial genome annotation rate is 87.8% compared to 72.9% with Bakta, while Baktfold's median bacterial annotation rate of remaining hypothetical proteins is 50.1% (n=290258). For archaea, Baktfold's overall median annotation rate is 71.5% compared to Prokka's 35.8%, with a median archaeal annotation rate of hypothetical proteins of 68.0% (n=14058), making Baktfold the most sensitive automated archaeal annotation method by far. Baktfold is implemented in Python 3 and runs on MacOS and Linux systems. It is freely available under a MIT license at https://github.com/gbouras13/baktfold.
|
|
Scooped by
mhryu@live.com
April 1, 11:28 PM
|
Plasmid conjugation is central to plasmid maintenance and spread among bacteria. Conjugation assays in liquid or on solid agar media are commonly used to quantify plasmid conjugation rates. Plasmids with short, rigid conjugative pili are thought to conjugate more efficiently on surfaces, whereas plasmids encoding long, flexible pili can conjugate efficiently in liquid medium. However, this pattern has not been tested systematically. Here, we perform standardized conjugation assays on a collection of 13 conjugative plasmids belonging to families that play a key role in AMR transmission and encode different conjugative pili types. We confirm that only the plasmids encoding long flexible pili conjugate efficiently in liquid. Furthermore, most transconjugants that arise from liquid assays involving plasmids with short, rigid pili can be attributed to transfer happening after the assay itself, on the surface of selective plates. This effect is amplified when using auxotrophic rather than antibiotic resistance markers, and impacts measures of transfer and defence efficiency. Finally, most of the tested plasmids with short pili had very high conjugation rates on surfaces, suggesting their transfer is mostly limited by physical constraints.
|
|
Scooped by
mhryu@live.com
April 1, 11:07 PM
|
Horizontal gene transfer plays a key role in bacterial evolution, yet its efficiency under natural conditions, especially between genetically distinct strains, remains unclear. Using Staphylococcus aureus as a model, we found that gene transfer via various mechanisms is significantly restricted between strains from different clonal complexes (CCs), with the notable exception of lateral transduction, which occurs at high frequency. Interestingly, some strains exhibited a promiscuous ability to accept diverse mobile genetic elements. These strains were defective in key immune defences, specifically the Type I restriction-modification systems that normally protect against foreign DNA. A broader analysis revealed that such immune-deficient mutants are widespread within S. aureus populations. Our study uncovered a trade-off that may account for their persistence in nature: although these mutants are more susceptible to phage attack, they gain an evolutionary advantage by acquiring new genes - such as those conferring antibiotic resistance - which would enhance survival under selective pressure. These immune-deficient cells act as gateways for foreign DNA, which, once integrated and advantageous, can spread within the same CC. Our findings highlight the role of immune-deficient bacteria in facilitating the emergence of novel virulence factors and antibiotic resistance, emphasising their importance in shaping bacterial evolution. The efficiency of horizontal gene transfer between different bacterial lineages is often unclear. Here, Figueroa et al. show that lateral transduction is the primary driver of gene exchange between Staphylococcus aureus lineages, and immune-deficient mutants enable the spread of new genetic trait
|
|
Scooped by
mhryu@live.com
April 1, 10:39 PM
|
Pseudomonas putida is a gram-negative bacterial species increasingly utilized in biotechnology due to its robust growth, ability to degrade aromatic compounds, solvent tolerance, and genetic tractability. In this study, we report a comprehensive multi-strain analysis of 164 P. putida strains based on the reconstruction of a pan-putida metabolic network and the formulation of strain-specific genome-scale metabolic models (GEMs). We performed whole-genome sequencing and hybrid assembly for 40 strains, contributing a ~8% increase to the available genomic data for P. putida. Furthermore, high-throughput phenotypic profiling using the Biolog phenotype microarray system for 24 strains on 190 unique carbon sources, along with 15 aromatic compounds not present on Biolog plates, yielded 4,920 unique strain-phenotype measurements. These data were leveraged to curate GEMs for 24 representative strains, including a refined model for strain KT2440, which comprised 1,480 genes and 2,191 metabolites, achieving a prediction accuracy of 91.2% in carbon utilization. Systematic comparison of genomes and GEMs revealed both conserved core pathways and significant allelic and functional divergence across strains, highlighting strain-specific variation in aromatic degradation. While pathways for protocatechuate and phenylacetate degradation were widely conserved, metabolic capabilities for compounds such as ferulate, phenol, and cresols varied markedly, suggesting adaptation to distinct ecological niches. Alleleome analysis of enzymes, such as PcaI and PcaJ, revealed distinct, functionally similar clades, indicating possible convergent evolution or horizontal gene transfer. These results provide computable resources and informative models for selecting P. putida strains with desired traits for biomanufacturing and bioremediation and offer insights into the evolution and phylogeny of the P. putida species.
|
|
Scooped by
mhryu@live.com
April 1, 5:14 PM
|
Quorum sensing (QS) enables bacteria to coordinate gene expression in response to population density, with LuxR-type transcription factors playing a central role in this process for many Gram-negative species. Traditionally understood as ligand-activated transcriptional regulators, LuxR-type proteins are increasingly recognized as targets of diverse protein-protein interactions (PPIs) that modulate their activity, stability, and specificity. This review synthesizes emerging insights into the regulatory landscape of LuxR-type receptors, focusing on direct PPIs that expand the functional repertoire of LuxR-type receptors. We classify LuxR-interacting partners into negative regulators, dual or context-dependent regulators, and global regulators, highlighting the well-characterized system of PqsE-RhlR in Pseudomonas aeruginosa as well as emerging candidates. We discuss how these interactions influence LuxR-type receptor outputs beyond canonical ligand-mediated gene regulation, through stabilization of active conformations, inhibition of dimerization, proteolytic regulation, and potential recruitment of transcriptional machinery. By examining functional parallels, structural determinants, and environmental integration, we highlight a framework in which LuxR-type receptors are subject to diverse regulatory PPI that allow for the fine-tuning of QS output. Understanding these regulatory mechanisms offers promising alternatives to conventional QS disruption strategies, for targeted interference with bacterial virulence and communication.
|
|
Scooped by
mhryu@live.com
April 1, 4:46 PM
|
In the evolutionary arms race between bacteria and bacteriophages, both parties have evolved diverse defense systems and counter-defense strategies. Protein phosphorylation is a ubiquitous regulatory mechanism that enables rapid cellular responses to internal and external stimuli. Accordingly, protein phosphorylation-based responses have been established in both the bacterial host and the infecting phages. This review provides an overview of protein Ser/Thr/Tyr kinases involved in bacterial defense and phage counter-defenses, with a particular focus on insights gained from mass spectrometry-based phosphoproteomic analyses of phage infections.
|
|
Scooped by
mhryu@live.com
April 1, 4:33 PM
|
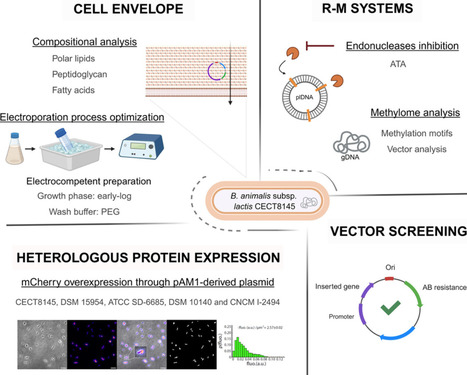
Bifidobacterium is a key genus of the gut microbiota. Among commercially used probiotics, Bifidobacterium animalis subsp. lactis CECT 8145 has been shown to exert health benefits in metabolic health and healthy aging. Remarkably, these effects persist in its postbiotic, heat-treated form, a feature attributed to its unique lipoteichoic acid composition. However, the challenges of genetically manipulating bifidobacteria hinder the identification of the molecular basis of this functionality. In this study, we report the first successful transformation of B. animalis subsp. lactis CECT 8145, alongside reproducible and high overexpression of heterologous recombinant proteins. Our approach integrates optimized electroporation protocols, tailored vector selection, and modulation of membrane fluidity, further enhanced by polyethylene glycol as a permeabilizing agent. Membrane fluidity - driven by the ratio of unsaturated to saturated fatty acids - is shown to be a quantifiable and tunable determinant of transformation efficiency. A comparative analysis of five B. animalis strains confirmed that transformation efficiency remains strongly strain-dependent, likely reflecting differences in ecological origin. Promoter pHUP yielded the highest heterologous protein expression levels, showing that careful promoter selection can substantially increase gene expression even in strains with low transformability, with protein expression levels increasing with plasmid copy number and reaching up to 104 arbitrary fluorescence units. Together, these findings establish a practical framework for the genetic engineering of recalcitrant, commensal probiotic strains such as CECT 8145. This work supports the development of next-generation probiotics and enables molecular validation of their health-promoting effects.
|
|
|
Scooped by
mhryu@live.com
Today, 5:09 PM
|
Microorganisms in extreme environments represent a promising source of novel metabolites, yet their global diversity and biosynthetic potential remain underexplored. Here, we reconstruct 78,213 bacterial and archaeal genomes from 2293 publicly available metagenomes and 3214 microbial isolates to establish a unified database, the Extreme Environment Microbiome Catalog (EEMC). The EEMC expands known global phylogenetic diversity, encompassing 32,715 representative species and nearly 4 billion non-redundant genes, 63.00% and 19.21% of which are previously unannotated, respectively. It also comprises 163,693 biosynthetic gene clusters, grouped into 64,733 gene cluster families, 58.68% of which are classified as novel, underscoring the functional diversity of microbial communities across various extreme habitats. We further develop protein large language models to predict genome-encoded candidate antimicrobial peptides (cAMPs) from the EEMC, identifying 3032 non-toxic candidates. Of 100 synthesized peptides, 84% demonstrate antibacterial activity, and all 50 tested cAMPs exhibit low cytotoxicity. Notably, six of the most potent cAMPs show significant efficacy against multidrug-resistant, Gram-negative pathogens in vitro, indicating their biomedical potential. Together, our study establishes the EEMC as a foundational resource for uncovering novel microbial lineages and biosynthetic capabilities, highlighting its substantial potential for drug discovery and laying the foundation for future advances in biotechnology and biomedicine. Extreme environments host diverse microbes, yet their global diversity remains underexplored. Here, the authors analyze both isolate genomes and metagenomes from various extreme habitats to construct a microbial genomic catalog, and use protein language models to identify antimicrobial peptides active against drug-resistant pathogens.
|
|
Scooped by
mhryu@live.com
Today, 4:54 PM
|
Pyrethroid insecticides are extensively applied owing to their potent insecticidal activity and low mammalian toxicity, yet their hydrophobicity results in persistent environmental residues. Here, we engineered a Bacillus subtilis spore surface display system to anchor Pseudomonas aeruginosa aminopeptidase (PaAps) on the spore coat and evaluated its potential for pyrethroid degradation. Surface-anchored PaAps efficiently degraded various pyrethroids, with β-cypermethrin showing the highest removal. The enzyme exhibited remarkable thermal stability and pH tolerance, with optimal activity at 60 °C and pH 8.0, along with enhanced long-term storage stability. Soil microcosm studies revealed that PaAps not only accelerated β-cypermethrin degradation but also influenced the indigenous microbial community to enhance bioremediation. This study demonstrates a robust and environmentally sustainable biocatalytic strategy for pyrethroid detoxification, with promising applications in environmental remediation and food safety.
|
|
Scooped by
mhryu@live.com
Today, 4:40 PM
|
Unspecific Peroxygenases (UPOs) are enzymes with significant potential as catalysts in synthetic chemistry as they catalyse selective oxygenation reactions on organic substrates at the expense of only hydrogen peroxide as the external oxidant. The demand for UPOs has stimulated considerable research into efficient heterologous expression systems for their production, which have included optimisation of the signal peptide (SP) included upstream of the UPO gene. In this study we report a comparison of the production of the prototypical and widely-used UPO variant from the fungus Agrocybe aegerita (rAaeUPO-PaDa-I-H) using both its native SP and variant SPs evolved for improved heterologous expression in yeast hosts. The results show that improvements in protein production identified using variant SPs in S. cerevisiae are not necessarily extended to the yeast Komagataella phaffii. Indeed in K. phaffii we observed a five to sevenfold improvement in the activity of crude secretates produced using the native fungal SP of AaeUPO, compared with SPs that were evolved for improved expression and screened in S. cerevisiae. These findings suggest that superior yields of this widely-used UPO can be obtained from scaled production in K. phaffii by using the native SP from the source fungus.
|
|
Scooped by
mhryu@live.com
Today, 4:17 PM
|
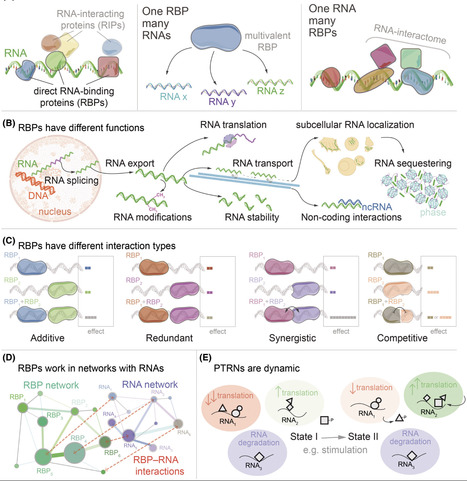
Post-transcriptional regulation of gene expression is orchestrated by RNA-binding proteins (RBPs), which regulate key aspects of the RNA life cycle including splicing, localization, translation, and decay. Although RBPs have been initially considered as isolated regulators, it is becoming clear that RNA molecules are commonly bound by several RBPs whose coordination directs their fate. These combinatorial interactions produce complex, context-dependent post-transcriptional regulatory networks (PTRNs) whose outcomes are difficult to predict. RBPs may also switch function depending on cell state, subcellular localization, or post-translational modification, adding further complexity to RNA regulation. This review focuses on recent technological advances expanding our ability to map and interpret PTRNs. Multiplexed methods allow profiling of the RNA-binding patterns of several RBPs in parallel, whereas deeper interaction proteomics studies reveal protein–protein connections and changes in distinct biological settings. Complementary RNA-targeting pulldown and single-molecule imaging strategies enable real-time and single-cell-resolution visualization of ribonucleoprotein assembly and dynamics, while functional high-throughput screens allow assignment of first order functions for these RBPs. Overall, these approaches set the stage for comprehensive decoding of the spatiotemporal structure of PTRNs and reveal how RBP interactions coordinate sets of RNAs to collectively regulate them in response to physiological demands. In addition to describing these systems-level approaches, we outline key future analytical and experimental innovations that could transform our understanding of RBP function. We believe that a systems-level understanding of RBPs as dynamic, integrated components of multiscale regulatory regimes is required to fully understand the complexity of gene expression control and its disruption in disease.
|
|
Scooped by
mhryu@live.com
Today, 3:03 PM
|
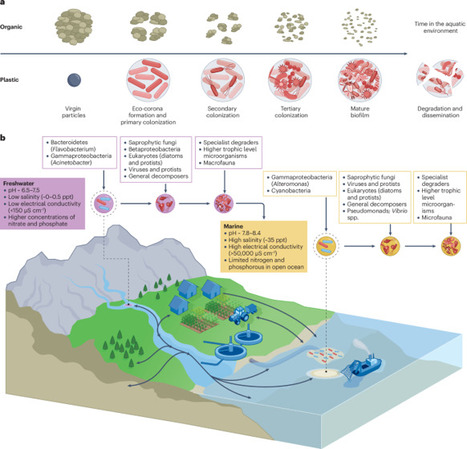
The escalating volume of plastic debris in the aquatic environment has created a novel ecological niche for microorganisms. This habitat, known as the plastisphere, hosts ecologically diverse microbial communities, yet its degree of divergence in composition and function from natural substrates remains a key topic of debate. In this Review, we provide a comprehensive overview of the aquatic plastisphere, highlighting its community composition, assembly dynamics and key functional traits, such as degradation pathways and nutrient cycling. We explore how this novel environment acts as a critical hub for the persistence and dissemination of human pathogens and antimicrobial resistance. We discuss how dynamic environmental factors, together with complex inter-kingdom metabolic interactions, can reshape community composition, accelerate the evolution of resistant pathotypes and influence pathogen virulence. We conclude by emphasizing that climate-induced changes in temperature, ultraviolet light radiation, salinity and hydrodynamic patterns are likely to further influence the microbial composition and phenotypic expression of pathogens within the aquatic plastisphere. Finally, we underscore the need for a One Health approach to study the plastisphere and advocate moving the field from descriptive ecology towards a mechanistic, policy-relevant framework to address the growing public health threat amid accelerating plastic pollution and climate-driven shifts. In this Review, Ormsby and Quilliam discuss how plastic pollutants create a critical, novel habitat for complex microbial communities, highlighting the community composition, assembly dynamics and key functional traits of the plastisphere. They explore the dual public health risks of pathogen dissemination and enrichment of antimicrobial-resistance determinants.
|
|
Scooped by
mhryu@live.com
April 1, 11:41 PM
|
Phase variation is viewed as a simple stochastic ON/OFF switch helping bacteria survive unpredictable environments. In minimal-genome pathogens like Mycoplasma bovis, simple sequence repeats (SSRs) introduce frameshifting InDels in key phase-variable genes, such as the Type III restriction-modification mod genes, typically assumed to result in binary expression. This study revisits this assumption using a heterologous E. coli system and single-cell mEGFP-based fluorescence profiling of M. bovis mod1 gene fragment containing SSR to determine if "OFF" states are truly silent. We find that the frameshifted construct remains active, showing low-level, heterogeneous expression in a frame-dependent manner, via frameshift suppression. This creates a range of expression rather than a strict binary switch. These findings suggest that phase-variable SSRs can function with upstream switches to form complex XNOR Boolean logic gates. This demonstrates that sophisticated logic gates can emerge directly from coding sequence architecture enhancing diversity and adaptability to promote evolutionary resilience in compact bacterial genomes.
|
|
Scooped by
mhryu@live.com
April 1, 11:32 PM
|
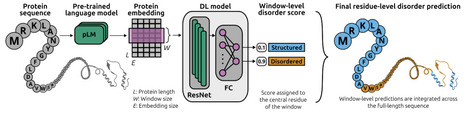
Intrinsically disordered proteins (IDPs) play an important role in a wide range of biological functions and are linked to several diseases. Due to technical difficulties and the high cost of experimental determination of disorder in proteins, combined with the exponential increase of unannotated protein sequences, the development of computational methods for disorder prediction became an active area of research in the last few decades. In this work, we present emb2dis, a deep learning model that uses protein language models (pLMs) to predict disorder from sequence. The emb2dis tool is a pre-trained model that receives as input a protein sequence, calculates its pLM embedding and passes it to a deep learning model. In contrast to existing approaches, emb2dis integrates informative sequence representations with a novel architecture that combines residual networks (ResNets) and dilated convolutions. This design effectively enlarges the receptive field of the convolution operation, enabling the model to better capture an extended context of each amino acid. At the output, emb2dis assigns a disorder propensity score to each residue in the sequence. The model was evaluated on datasets from the latest CAID3 blind benchmark for disorder prediction, where it achieved first place in the Disorder-PDB category, exhibiting strong performance with high AUC and Fmax scores. Additionally, it ranked among the top ten methods on the Disorder-NOX dataset. We provide a freely available web-demo for emb2dis and a source code repository for local installation. Weblink for the tool: https://sinc.unl.edu.ar/web-demo/emb2dis/
|
|
Scooped by
mhryu@live.com
April 1, 11:22 PM
|
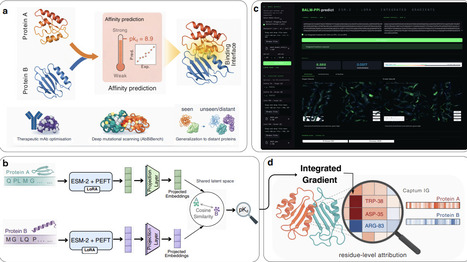
Predicting protein-protein binding affinity from sequence alone remains a bottleneck for antibody optimization, biologics design and large-scale affinity modelling. Structure-based methods achieve high accuracy but cannot scale when complex structures are unavailable. Here we present a framework that reframes affinity prediction as metric learning: two proteins are projected into a shared latent space in which cosine similarity directly correlates with experimental binding affinity, and the protein language model encoder is adapted through parameter-efficient fine-tuning (PEFT). On the PPB-Affinity benchmark, the model achieves Pearson r = 0.89 on a random split, generalises to evolutionarily distant proteins (r = 0.61 at <30% sequence identity) and surpasses structure-based deep learning baselines across biological subgroups, without any three-dimensional input. On the strictly de-overlapped AB-Bind dataset, few-shot adaptation with 30% of assay data (Pearson r = 0.756, RMSE = 0.688) outperforms methods trained on 90% of data; consistent gains are observed across nine diverse AbBiBench deep-mutational-scanning assays with 10-30% labelled variants. Residue-level explainability reveals that the model concentrates importance on interface-localised residues aligned with experimentally validated interaction hotspots across enzyme-inhibitor, and antibody-antigen systems. Together, these results establish a scalable, explainable and data-efficient route to protein-protein binding affinity prediction and therapeutic antibody optimisation from sequence alone.
|
|
Scooped by
mhryu@live.com
April 1, 10:40 PM
|
Nicotine, tobacco’s addictive and potent insecticidal alkaloid, has shaped human history, agriculture, and the plants that produce it. However, the enzymatic steps and reaction mechanisms involved in nicotine biosynthesis remain elusive. Here, we reveal that the final coupling reaction is stabilized by glycosylation via a uridine diphosphate (UDP)-glycosyltransferase, reduced and activated by an A622, condensed through a stereoselective intermolecular Mannich-like reaction, sequentially oxidized by a berberine bridge enzyme-like (BBL), and finally deglycosylated by a β-glucosidase to yield nicotine. A 5-component metabolon assembles at vacuolar membranes to channel both nicotine biosynthesis and its transport. We reconstituted this metabolon both in vitro and heterologously in vivo. Abrogating any of these components depletes nicotine accumulations. A multidrug and toxic compound extrusion (MATE) transporter is essential for efficiently engineering nicotine production in heterologous plant species, which confers pest resistance. This work completes the nicotine biosynthesis pathway and provides critical insights into the intermolecular Mannich-like reaction, a fundamental mechanism for scaffold formation in many plant alkaloids.
|
|
Scooped by
mhryu@live.com
April 1, 10:17 PM
|
Pseudomonas putida is a gram-negative bacterial species increasingly utilized in biotechnology due to its robust growth, ability to degrade aromatic compounds, solvent tolerance, and genetic tractability. In this study, we report a comprehensive multi-strain analysis of 164 P. putida strains based on the reconstruction of a pan-putida metabolic network and the formulation of strain-specific genome-scale metabolic models (GEMs). We performed whole-genome sequencing and hybrid assembly for 40 strains, contributing a ~8% increase to the available genomic data for P. putida. Furthermore, high-throughput phenotypic profiling using the Biolog phenotype microarray system for 24 strains on 190 unique carbon sources, along with 15 aromatic compounds not present on Biolog plates, yielded 4,920 unique strain-phenotype measurements. These data were leveraged to curate GEMs for 24 representative strains, including a refined model for strain KT2440, which comprised 1,480 genes and 2,191 metabolites, achieving a prediction accuracy of 91.2% in carbon utilization. Systematic comparison of genomes and GEMs revealed both conserved core pathways and significant allelic and functional divergence across strains, highlighting strain-specific variation in aromatic degradation. While pathways for protocatechuate and phenylacetate degradation were widely conserved, metabolic capabilities for compounds such as ferulate, phenol, and cresols varied markedly, suggesting adaptation to distinct ecological niches. Alleleome analysis of enzymes, such as PcaI and PcaJ, revealed distinct, functionally similar clades, indicating possible convergent evolution or horizontal gene transfer. These results provide computable resources and informative models for selecting P. putida strains with desired traits for biomanufacturing and bioremediation and offer insights into the evolution and phylogeny of the P. putida species.
|
|
Scooped by
mhryu@live.com
April 1, 5:02 PM
|
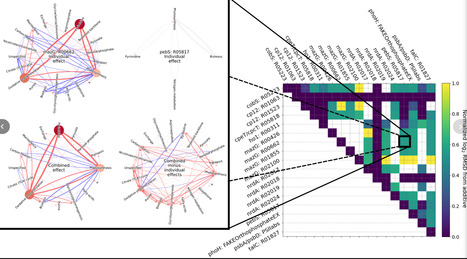
Bacteriophage auxiliary metabolic genes (AMGs) alter host metabolism by hijacking reactions, but previous studies mostly inferred their roles from annotations, ignoring system-wide impacts and phage production. Here we integrate AMGs and phage assembly into a genome-scale metabolic model of Prochloroccocus marinus MED4 infected by P-HM2. We show that 17 directly hijacked reactions substantially affect more than 30% of the reactions in MED4 metabolism, including carbon fixation, photosynthesis, and nucleotide synthesis, distinguishing these AMGs as either phage aligned—shifting feasible reaction velocities in accordance with maximal phage production—or phage antialigned. Pareto optimization reveals that phage-aligned reactions alter phage-host growth trade-offs, while phage-antialigned reactions do not. We experimentally validate our predictions of system-level AMG impacts by measuring the N-dependent effect of P-HM2 cp12 expression on growth in a model relative of the genetically intractable MED4, Synechococcus elongatus. We also show that AMGs’ indirect impacts are synergistically and antagonistically coupled, providing systems-level insight into AMG perturbations and highlighting how nontrivial cascading effects shape host metabolism.
|
|
Scooped by
mhryu@live.com
April 1, 4:42 PM
|
Waste management aboard the International Space Station is a critical challenge due to limited space, microgravity, and continuous waste generation. This manuscript explores biotechnological strategies that convert waste into valuable resources, supporting sustainability and reducing reliance on Earth. Biowaste decomposition transforms organic waste into nutrients and biogas, while regenerative life support systems recycle carbon, water, and minerals. Biomining enables microbes to extract rare elements and upcycle electronic waste into mission-critical metals. Engineered extremophiles demonstrate resilience and productivity in space, supporting their potential role in regenerative life support systems. Biomanufacturing platforms that use alternative and in situ feedstocks demonstrated the capability of producing vitamins and pharmaceuticals from waste-derived feedstocks. These innovations enhance crew health, reduce launch mass, and offer scalable models for sustainable resource recovery, marking a shift toward self-sufficient extraterrestrial habitats while simultaneously establishing the foundations for materially closed-loop bioregenerative life-support systems essential for Mars missions and long-term off-Earth settlements.
|


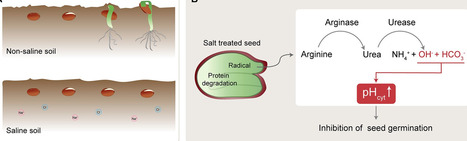






















https://elifesciences.org/articles/101732 (A) In non-saline soil conditions (top), seeds (brown circles) germinate and grow to become seedlings (green). In saline soil (bottom), which contains more sodium (Na+; pink) and chlorine (Cl-; blue) ions, seed germination is inhibited. (B) Under salt stress conditions, protein degradation in the seed creates arginine, which is converted to urea by arginase. The urea is subsequently hydrolyzed by urease, resulting in increased levels of ammonium ions (NH4+), bicarbonate ions (HCO3-), and hydroxide ions (OH-). The bicarbonate ions and hydroxide ions increase the cytoplasmic pH (pHcyt), thereby inhibiting seed germination.