Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:43 PM
|
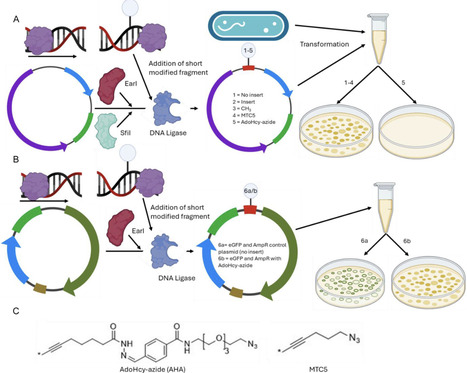
Artificial control of gene expression in bacteria offers interesting prospects for influencing bacterial pathogenicity and antibiotic resistance. We show that the methyl-transferase cofactor, AdoHcy azide, can silence gene expression in modified plasmids in some strains of E. coli, where ampicillin and kanamycin resistance as well as eGFP genes were selectively and independently disabled. The disabling of transcription is likely due to steric inhibition during transcription initiation, which is supported by Sanger and nanopore sequencing results. Both sequencing methods showed that 3–6 nucleotides were absent from around the modification site. Postgrowth, extracted AmpR/eGFP plasmid shows evidence of restriction, with sections of the plasmid, including the modification site, missing for the AdoHcy azide modified plasmids. Notably, the AdoHcy azide modification on the DNA appears to be resistant against demethylation in the BL21 strain of E. coli.
|
|
Scooped by
mhryu@live.com
Today, 10:11 PM
|
The biological activity of many proteins is influenced by glycosylation, underscoring its essential role. However, no established recombinant technique currently enables the controlled shedading of glycosylated extracellular loops from transmembrane proteins. Here, we describe enzymatically controlled release of displayed proteins and peptides (ENCOREP), a strategy that enablesin situ expression at the cell surface and protease-mediated release, followed by collection from the cell culture medium. Using ENCOREP, we achieved the production of glycosylated peptides. These glycopeptides, derived from the large extracellular loop of the highly glycosylated CD63, illustrate the type of targets that are otherwise inaccessible with current methods but can be readily obtained using ENCOREP. Overall, ENCOREP provides a rapid and reliable approach to obtain glycosylated proteins or peptides while bypassing the conventional signal peptide–dependent secretory pathway.
|
|
Scooped by
mhryu@live.com
Today, 6:43 PM
|
Gene regulation through promoter engineering is a cornerstone of synthetic biology, enabling precise control over transcriptional networks. However, experimental approaches remain labor-intensive. While artificial neural networks (ANNs) have improved regulatory element prediction, tools for promoter–transcription factor binding site (TFBS) recombination are still lacking. We present an ANN framework for context-aware design of synthetic promoters in Saccharomyces cerevisiae. The model predicts optimal TFBS insertion sites and the extent of promoter rewriting needed for successful integration. Applying this, we screened 6,011 native yeast promoters for compatibility with the TetR TFBS, generating a ranked list of high-confidence promoter–TFBS pairs. Experimental validation showed that model-designed promoters achieved repression rates up to 98.4%, without prior experimental characterization or tuning. We further rewired the yeast transcriptional network by introducing glucose-dependent regulation of an essential gene via Mig1 TFBS insertion. These results establish a scalable, predictive method for engineering regulatory sequences and reprogramming transcriptional logic.
|
|
Scooped by
mhryu@live.com
Today, 2:33 PM
|
In E. coli, chromosome replication is regulated through ATP/ADP state of the DnaA initiator. The DDAH system inactivates DnaA in the post-initiation stage by promoting ATP hydrolysis through timely binding of the DNA-bending protein IHF to the datA locus, while the DARS2 locus reactivates DnaA in the pre-initiation stage via binding of IHF and another nucleoid protein Fis. The iron-sulfur cluster [(Fe-S)] assembly factor YgfZ is known to sustain replication initiation, central carbon metabolism, redox state and modification of tRNA A37 residues by MiaB, but the link between initiation and the others remains unclear. This study shows that YgfZ regulates initiation primarily by downregulating the DDAH system by repressing datA-IHF binding in a manner independent of MiaB. Also, the [Fe-S]-binding protein MnmA moderately downregulates datA-IHF binding. Furthermore, YgfZ globally downregulates basal IHF binding across the genome, while preserving IHF's timely binding at key loci including oriC and datA during the cell cycle, highlighting a novel strategy: YgfZ modulates both the cellular metabolic states and global genome dynamics to control replication initiation under various growth conditions.
|
|
Scooped by
mhryu@live.com
Today, 1:53 PM
|
Xyloglucan oligosaccharides (XyGOs) are emerging prebiotics with limited sustainable production strategies. Here, two endoxyloglucanases, XyGH5 and XyGH74, from Paenibacillus sp. XP01 were identified. Genetic knockout revealed their synergistic role in bacterial growth on xyloglucan. Biochemical characterization showed that XyGH74 exhibited superior activity (Vmax = 76.25 U/mg) and thermal stability (optimum at 70 °C) compared to XyGH5, with its carbohydrate-binding modules (CBMs) crucially enhancing catalytic efficiency and substrate affinity. We further employed the highly efficient XyGH74 to produce tamarind xyloglucan oligosaccharides (TXOS). In vitro fermentation with human gut microbiota demonstrated that TXOS significantly modulated microbial composition, enriching beneficial genera such as Bacteroides and Parabacteroides, and notably enhanced the production of short-chain fatty acids, particularly butyrate, compared to the native polysaccharide. This study provides direct genetic evidence for synergistic glycoside hydrolase function in vivo and establishes XyGH74 as a robust enzymatic tool for scalable, high-value prebiotic production with enhanced health benefits.
|
|
Scooped by
mhryu@live.com
Today, 1:47 PM
|
A miniaturized variant of the artificial luciferase (ALuc), named picALuc, has been generated through the deletion of N- and C-terminal residues in ALuc. Although picALuc is small and active, questions remain regarding its the structural organization and inter-residue interactions in the protein. Here, combining computational analysis and mutational studies, we show that the E50A mutation in picALuc results in an increased bioluminescence activity of the protein. Specifically, we generated a structural model of picALuc using the available structure of the Gaussia luciferase (GLuc) that revealed a ‘hole’ in the structure due to the deletion of N-terminal α-helices. Gaussian-accelerated molecular dynamics (GaMD) simulation revealed a rapid ‘compaction’ of the picALuc structure during the initial phase of the simulation and a number of residues such as E10, E50, and D94 showed salt bridge interactions. Mutation of the residues E10, E50, and D94 individually to an A revealed increased bioluminescence activity of the E50A mutant, while E10A and D94A mutants showed activities similar to the WT protein in living cells. In vitro assays revealed an increase in the Vmax of the E50A mutant, while Khalf and thermal stability of the mutant remained unchanged. Further, dynamic cross-correlation and principal component analyses of the GaMD simulation trajectories of the WT and the E50A mutant picALuc revealed altered collective dynamics in the protein. Finally, we developed a protein fragment complementation assay using picALuc that allows for the monitoring protein–protein interactions (PPIs) in live cells. We envisage that the brighter picALuc reported here will find broad applicability in developing bioluminescence-based assays.
|
|
Scooped by
mhryu@live.com
Today, 1:32 PM
|
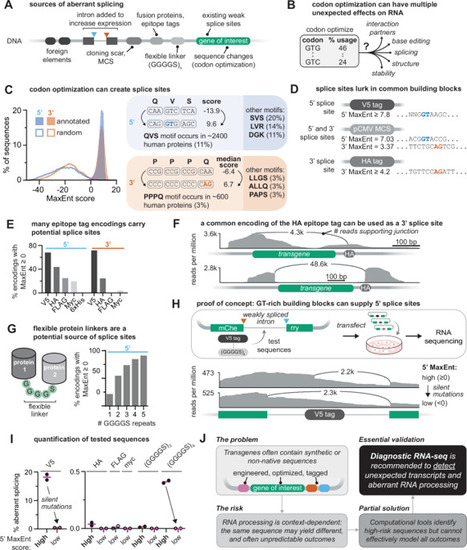
Plasmids are the workhorses of molecular biology: fast, flexible, and often taken for granted. We clone, overexpress, tag, and mutate freely, assuming they will faithfully produce RNA transcripts that match the intended DNA sequence. This assumption is rarely tested and often invalidated. Sequences in plasmid backbones, epitope tags, and codon-optimized regions may inadvertently harbor cryptic promoters or splice sites. The resulting unexpected transcripts and proteins, while often undetected, can distort results and propagate false conclusions through papers, grants, and even clinical trials. In this perspective, we highlight published cases where plasmids have distorted results and misled interpretation. We examine the mechanisms and consequences of plasmid-associated expression artifacts and offer practical strategies to minimize them. Finally, we call for a revision of community standards for experiments using transgenes: deposit complete plasmid sequences and verify the resulting transcripts using RNA-seq.
|
|
Scooped by
mhryu@live.com
Today, 1:23 PM
|
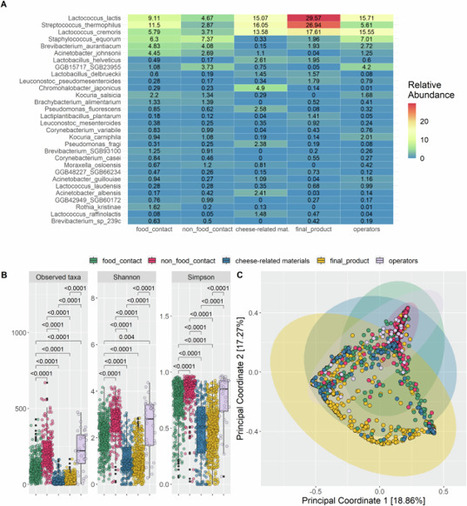
Metagenome-assembled genomes (MAGs) provide crucial insights into the genomic diversity of uncultured microbes. However, MAG datasets deposited in public repositories such as INSDC are often difficult to reuse due to heterogeneous quality, inconsistent taxonomic and functional annotations, and insufficiently curated environmental metadata. While secondary MAG databases such as MGnify, IMG/M, and SPIRE provide standardized resources, they reconstruct MAGs de novo from public metagenomic reads and therefore do not represent the original MAGs reported in publications. To address this gap, we developed Microbiome Datahub, an open-access platform that systematically aggregates and re-annotates original MAGs from INSDC. We collected 214,427 MAGs, predicted genes by DFAST, performed quality assessment with CheckM, standardized taxonomic assignments with GTDB-Tk, inferred 27 phenotypic traits using Bac2Feature, assigned proteins to MBGD ortholog clusters and KEGG Orthology IDs using PZLAST, and annotated environmental metadata with the Metagenome and Microbes Environmental Ontology. Across these MAGs, the average completeness was 80.5% and contamination 1.8%; notably, the most frequent values were >95% completeness and <1% contamination, indicating that the majority of MAGs are of high quality. Comparative analyses showed that Microbiome Datahub provides phylogenetically and environmentally diverse MAGs: while the majority originated from vertebrate gut environments, a substantial number were also recovered from other habitats such as groundwater, including nearly 10,000 MAGs from the Patescibacteria. Inference of 27 phenotypic traits, including optimum growth temperature, further revealed ecological differentiation across phyla. Protein clustering revealed 56 million identity 40% clusters, with the majority unique compared with MGnify and GlobDB, and ~19% of proteins unassigned to MBGD ortholog clusters, underscoring their novelty. Microbiome Datahub integrates MAG genome sequences, gene and protein predictions, quality metrics, environmental and taxonomic annotations, ortholog cluster assignments, and phenotype predictions, all accessible via a web interface, API, and bulk downloads. By combining original MAGs with curated metadata and functional annotations, Microbiome Datahub constitutes a comprehensive and reusable resource that will accelerate microbiome and microbial genomics research. Video Abstract
|
|
Scooped by
mhryu@live.com
Today, 1:06 PM
|
Microbial resources are crucial for biotechnology development and fundamental scientific research. Traditional microbial techniques fail to isolate and cultivate the vast majority of microorganisms in nature, severely limiting the discovery of novel microbial resources. The rise in artificial intelligence (AI) technologies provides new computational tools to overcome bottlenecks in microbial resource discovery and utilization. This review comprehensively examines the development of AI technologies in microbial isolation and cultivation over the past three decades from the perspective of microbial resource discovery. We propose a five-stage framework: the germination period (1997–2008), the early exploration period (2008–2015), the rapid development period (2015–2019), the deep learning (DL) explosion period (2020–2022), and the AI integration period (2023–present). We focus on how AI technologies at each stage address core challenges in microbiology—including insufficient knowledge reserves, dynamic phenotypic changes, and complex cultivation conditions—through applications at the genome, individual, and community levels. Our analysis demonstrates that, as AI technologies advance iteratively, microbial isolation and cultivation methods are transitioning from experience-driven to data-driven approaches, from single-objective to systematic integration, and from passive screening to active design. This methodological transition is expanding the scope of microbial resource discovery.
|
|
Scooped by
mhryu@live.com
Today, 9:37 AM
|
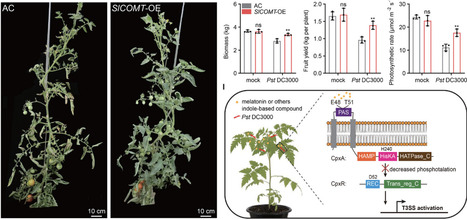
In defending against pathogens, plants deploy diverse secondary metabolites and signaling molecules. Among these, melatonin orchestrates plant growth and development, modulates stress responses, and regulates intracellular redox homeostasis and signaling. However, the mechanisms of melatonin in plant-pathogen interaction are rarely reported. Using Pseudomonas syringae pv. tomato DC3000 (Pst DC3000) as model bacteria, we designed a two-step high-throughput screening strategy to screen the plant natural product library and the bacterial mutant library. This study reveals that melatonin is perceived by a bacterial receptor histidine kinase CpxA, which subsequently modulates bacterial virulence. In detail, bacterial CpxA senses melatonin through Glu48 and Thr51 sites located in the periplasmic sensor region. Thus, melatonin inhibits autophosphorylation of CpxA and decreases transphosphorylation of the response regulator CpxR. The DNA-binding capacity of CpxR to promoters of type III secretion system (T3SS) genes is weakened by reduced phosphorylation cascade of CpxA/R, inhibiting bacterial T3SS genes expression and virulence. We also showed that increasing melatonin synthesis in plants can enhance disease resistance and sustain crop productivity. This study illustrates a previously unknown mechanism by which plants disarm the pathogenicity of bacteria, as well as provide effective molecular targets for crop genetic improvement and biopesticides development.
|
|
Scooped by
mhryu@live.com
Today, 2:00 AM
|
Bacteriocins are ribosomally synthesized antimicrobial peptides produced by various classes of bacteria, exhibiting broad-spectrum activity that makes them promising candidates for applications in food preservation and medicine. Their inherent stability under extreme pH, temperature, and salinity conditions further supports their functional versatility. However, the widespread industrial application of bacteriocins remains constrained by the low titres typically achieved during fermentation. Despite extensive efforts to optimize production using batch and fed-batch fermentation strategies, the resulting titres remain inadequate for economically viable large-scale manufacturing. This review aims to provide a comprehensive overview of novel process strategies developed over the past two decades to enhance bacteriocin yields during fermentation. One of the primary challenges is the inhibition of microbial growth due to the accumulation of lactic acid or the bacteriocin itself during production. To address this, in-situ product removal techniques—such as co-cultivation with lactic acid-consuming microorganisms, in-situ adsorption, filtration, and foam fractionation—have been explored, yielding notable improvements in bacteriocin titres. Additionally, stress-induced production strategies involving biological (e.g. co-culture with competing microbes), chemical (e.g. salinity and pH stress), and physical (e.g. agitation, temperature, and aeration stress) stimuli have also demonstrated success in enhancing bacteriocin synthesis. This review underscores the importance of these innovative fermentation approaches and highlights the need for further research focused on scaling up such processes. Advancing these strategies is critical to realizing the full potential of bacteriocins in food safety, antimicrobial therapy, and broader biomedical applications.
|
|
Scooped by
mhryu@live.com
Today, 1:52 AM
|
Chemical defenses are fundamental in organismal biology and widely used in medicine and agriculture. Plant defense chemistry evolves in response to various selective pressures, particularly herbivory, and theory has emphasized predicting toxin abundance and diversity. Here we test hypotheses about the evolution of structural innovation in chemical defense by combining molecular complexity metrics, metabolomics, molecular docking, and phylogenetic analyses, using milkweed cardenolides, steroidal glycosides that inhibit animal Na+/K+-ATPases. We identify the addition of a nitrogen–sulfur (N,S) heterocycle in highly substituted cardenolides as a major structural innovation that restores toxicity against coevolved natural enemies, such as the monarch butterfly. This toxicity is likely achieved by rigidifying the cardenolide scaffold and creating additional nonelectrostatic interactions within the Na+/K+-ATPase binding pocket, thereby enhancing binding affinity despite target-site resistance. Two biosynthetically distinct N,S-cardenolides, uscharin and labriformin, rank among the most complex structures in this chemical class and show divergent macroevolutionary histories: uscharin represents an ancestral character state with repeated losses, whereas labriformin has independently evolved multiple times in later-diverging lineages. This pattern across Asclepiadoideae indicates that the structural innovation evolved repeatedly, apparently limited by lineage-specific biosynthetic constraints among precursor pathways. N,S-cardenolides occur in over 75% of the 59 Asclepias species examined here, and species producing N,S-cardenolides exhibit greater cardenolide abundance, richness, metabolomic space, and toxicity against adapted organisms. More generally, structural innovation defines a distinct evolutionary axis in plant chemistry, enabling defense diversification and adaptive recovery of toxicity. Such innovations are predicted to build on existing molecular scaffolds in response to ecological challenges, here driven by coevolving specialist herbivores.
|
|
Scooped by
mhryu@live.com
Today, 1:44 AM
|
Resolving the biological and geological events that led to the origin of eukaryotes is an ongoing challenge in biology. A major step in the evolution of complex cellular life was the merger between an ancestral host cell and a bacterium (that became the mitochondrion) some two billion years ago. Recently, metagenomics has enabled the reconstruction of a broad diversity of genomes, referred to as the Asgard Archaea. The Asgards are monophyletic with eukaryotes on the tree of life. Asgards have an array of genes, previously thought exclusive to eukaryotes, involved in cellular trafficking, the ubiquitin system, endosomal sorting, and cytoskeleton formation, with growing evidence demonstrating the functions of these proteins mirror those in eukaryotes. This gene repertoire suggests that these Archaea are descendants of the archaeal host from which eukaryotes evolved. Increased sampling has revealed that Asgard lineages are metabolically versatile and play key roles in various ecosystems and uncovered evolutionary transitions between Archaea and eukaryotes, such as innovations in eukaryotic defense systems. The positioning of eukaryotes in the Asgards is debated, but eukaryotes appear to branch within the Heimdallarchaeia. Lineages within this group, particularly Hodarchaeales and Kariarchaeaceae, contain a broad repertoire of eukaryote-like traits, including high-energy yielding metabolisms. Observing and studying Asgard interactions with bacterial descendants of mitochondria in a modern setting will transform our understanding of the origin of complex cellular life.
|
|
|
Scooped by
mhryu@live.com
Today, 11:14 PM
|
For the first time, the AlphaFold protein-structure database will include predictions of complexes of proteins — with the addition of 1.7 million ‘homodimers’ comprising two interacting strands of the same molecule. Since its release in 2021, this repository has become a bedrock in discovery and a first port of call for research projects that try to understand life at the molecular level. In the coming weeks, the AlphaFold database will also include complexes called heterodimers, which are made of two different proteins.
|
|
Scooped by
mhryu@live.com
Today, 6:51 PM
|
Diverse phage-bacteria communities coexist at high densities in environmental, agricultural, and human-associated microbiomes. Phage-bacteria coexistence is often attributed to coevolutionary processes mediated by complex, pairwise infection networks. Here, using in vitro experiments and mathematical models, we explore how higher-order interactions function as a complementary, ecological feedback mechanism to stabilize phage-bacteria communities. To do so, we examine an environmentally-derived, synthetic phage-bacteria community comprised of five marine heterotrophic bacteria (Cellulophaga baltica and Pseudoalteromonas strains) and five associated phage. We used Bayesian inference to reconstruct free phage production in one-step growth experiments and then forecasted pairwise phage-bacteria community dynamics over multiple infection cycles. In contrast to model predictions of rapid bacterial population collapse, each bacterial strain persisted in the community. We hypothesized and then experimentally validated the relevance of infection attenuation at relatively high viral densities. We extended models into a community context, corroborating complex coexistence of all phage and bacteria. Life history traits inferred in community fits often differed from those inferred in a pairwise context, implicating higher-order interactions as an additional, ecological stabilization mechanism. Follow-up experiments confirm that phage traits (including burst size) can shift when infecting single vs. multiple strains. More broadly, these findings suggest that complex community coexistence of phage and bacteria may be more common than anticipated when including feedback mechanisms outside of the growth-dominated regimes of fitted pairwise models that do not reflect the full scope of ecologically relevant contexts.
|
|
Scooped by
mhryu@live.com
Today, 2:36 PM
|
Colonization of plastic surfaces by microbial biofilms offers a promising starting point for engineering efficient biodegradation systems. However, most studies to date focus on characterization or prevention of biofilms on plastics in diverse environments and the potential biotechnological application for these systems has been underexplored. To address this, we report the efficient adhesion of E. coli cells to a range of plastic surfaces through overexpression of two key determinants of bacterial biofilm formation; curli and Antigen 43 (Ag43). A general trend of higher total biomass was observed from curli-mediated adhesion, but more uniform adhesion from Ag43 overexpression. We further demonstrate application of this technology through inducible adhesion of E. coli to polyethylene terephthalate (PET) surfaces and concurrent secretion of the PET depolymerase PHL7. Co-overexpression of curli fibres and secreted PHL7 resulted in 5.6-fold increase in terephthalic acid release in comparison to the non-adherent control. These methods offer a general approach to programmable adhesion of genetically tractable cells to plastic surfaces and concurrent secretion of degradative enzymes, and are anticipated to be broadly applicable across the field of plastic bioremediation technologies.
|
|
Scooped by
mhryu@live.com
Today, 1:58 PM
|
High-throughput sequencing has generated vast genomic repositories that remain under-annotated, hampering enzyme discovery. We present an integrated pipeline that (i) builds a high-resolution, cross-kingdom phylogenetic database, (ii) mines candidates via multilocus phylogeny, (iii) predicts activities using an evolutionary-scale protein language model, and (iv) removes false positives through multilevel residue–atom contact rescoring. When applied to the r-BOX pathway, this approach uncovered numerous previously undocumented FadB, BktB, Ter, and YdiI homologues. Our activity model achieved R2 = 0.68 and reduced the RMSE on high-value targets by 11% compared to the prior SOTA (UniKP). Contact scoring improved early enrichment (EF1%) by 16-fold. Experimental validation targeting FadB increased titers from 0.65 g/L (shake flasks) to 1.7 g/L, reaching 10.2 g/L in a fermentation process. Together, these results establish a robust, generalizable framework for discovering scarce, high-value enzymes and prioritizing functional variants at scale.
|
|
Scooped by
mhryu@live.com
Today, 1:50 PM
|
Lactopontin (LPN) plays a critical role in the growth and development of infants. Caprine milk and bovine milk are the primary raw materials for infant formulas. However, the differences between caprine and bovine LPN have not been studied. In this study, the structure, digestive characteristics, and active peptide composition of LPN from different sources were compared. Two N-glycosylation sites, Asn101 and Asn208, were identified in bovine LPN, while a single N-glycosylation site, Asn79, was found in caprine LPN. The in vitro infant digestion simulation results indicated that caprine LPN released a greater quantity of small peptides and amino acids. The intestinal digestion products were subsequently analyzed. The digestive peptides derived from caprine LPN may possess various potential biological functions. These findings provide insights into optimizing protein digestion and nutrient absorption in infant formula.
|
|
Scooped by
mhryu@live.com
Today, 1:39 PM
|
Type III CRISPR–Cas systems confer antiviral immunity via cyclic oligoadenylate (cOA) signaling. Here, we elucidate a cooperative bacterial defense strategy involving two cOA-activated CRISPR-associated Rossmann fold (CARF)-containing effectors, adenosine deaminase CAAD and ribonuclease Csx1, in Thermoanaerobaculum aquaticum. Genomic analyses indicate widespread co-occurrence of CRISPR-associated adenosine deaminase (CAAD) with ancillary CARF-containing effectors in type III CRISPR systems, suggesting that multiple CARF-containing proteins may contribute to a coordinated cOA-dependent defense. Biochemical and structural studies reveal the intrinsic dynamics of CAAD hexamer, and demonstrate that cA4/cA6 binding stabilizes CAAD hexamers, triggering metal-ion-dependent conversion of ATP into inosine triphosphate. Concurrently, the downstream Csx1 is exclusively activated by cA4 to cleave single-stranded RNA. Strikingly, we found that both effectors are capable of degrading cA4, suggesting that this CAAD–Csx1 pair may be cross-regulated and achieve immunity through a dual-targeting mechanism: in response to infection, Csx1 degrades viral RNA while CAAD disrupts nucleotide metabolism via ATP deamination, which can be relieved via cA4 degradation when infection has been eliminated. This study proposes an enhanced defense mechanism through coordinated activation and regulation of multiple CRISPR effectors by a single signaling molecule, unveiling unprecedented complexity in CRISPR immunoregulation.
|
|
Scooped by
mhryu@live.com
Today, 1:29 PM
|
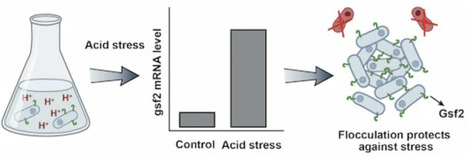
The social behaviors of microbes provide unique opportunities for testing social evolution theories. How can altruistic behaviors arise by natural selection is a central challenge in biology. Green-beard effect has been proposed as a basic mechanism for the evolution of altruistic behaviors. Yet, green-beard genes are generally thought to be rare. Here, we find that the Schizosaccharomyces pombe gsf2 gene mediates flocculation-like aggregation, and flocculation is triggered by acid stresses. gsf2-expressing cells preferentially adhere to each other. The expression of gsf2 is costly, but gsf2-expressing cells preferentially adhere to each other and protect each other from external stress. Gsf2 is highly variable in natural populations, likely contributing to different flocculation intensity. These findings suggest that gsf2 is a gradient green-beard gene that drives the altruism among gsf2 carriers. Moreover, we find that gsf2 is a new gene that originated very recently. Our results provide insights into the origin and evolution of green-beard genes.
|
|
Scooped by
mhryu@live.com
Today, 1:07 PM
|
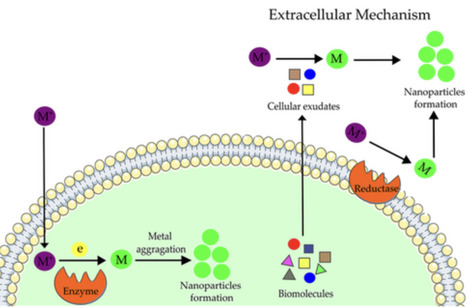
Biogenic nanoparticles are distinguished by their unique physical and chemical attributes, notably their potent antimicrobial activity against bacterial and fungal pathogens, as well as their cytotoxic effects on cancer cells. These nanoparticles are characterized by their biocompatibility, indicating their potential as effective antimicrobial agents and in oncological therapies. This article examines the existing literature on the antimicrobial and cytotoxic properties of nanoparticles derived from cyanobacteria, with particular emphasis on their implications for human health.
|
|
Scooped by
mhryu@live.com
Today, 11:03 AM
|
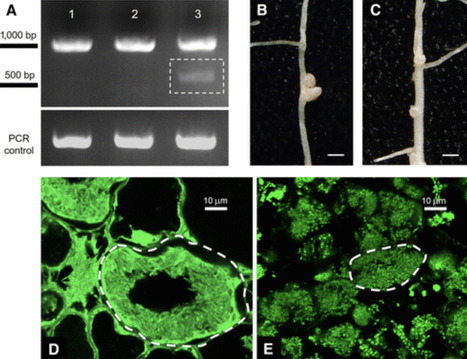
The model legume Medicago truncatula delivers nodule-specific cysteine-rich peptides to the intracellular bacteria within nodules to coerce the microbe into terminal differentiation, which coincides with nitrogen fixation in this species. Inside the host cell, the anterograde protein trafficking pathway is repurposed toward a new compartment, the symbiosome. Precise protein delivery within the nodule is critical to the success of the symbiosis in M. truncatula; without it, nodules form but do not fix nitrogen. For example, when the plant lacks DNF1, the nodule-specific 22-kDa subunit of the signal peptidase complex (SPC), the intracellular bacteria fail to fully differentiate, leading to defective nitrogen fixation. The present study shows that DNF1 became specialized in symbiosis through its nodule-specific expression, and we identified nodule-specific cis-elements that are crucial for that transcriptional control. Furthermore, we identified the nodule-specific SPC catalytic subunit and demonstrated that CRISPR/Cas9-induced mutation of this gene causes a symbiosis defect, which phenocopies the dnf1 mutant. These results suggest that a dedicated SPC in the nodule is co-opted for symbiosis through transcriptional regulation.
|
|
Scooped by
mhryu@live.com
Today, 9:30 AM
|
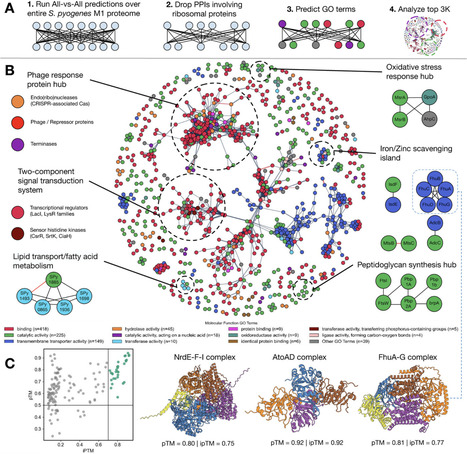
Protein–protein interactions (PPIs) are central to cellular processes and host-pathogen dynamics across all domains of life, yet comprehensive interactome mapping remains challenging at the proteome scale. Experimental approaches provide only partial coverage, while existing computational methods often lack generalizability across species or are too resource-intensive for large-scale screening. Here, we introduce ppIRIS (protein–protein Interaction Regression via Iterative Siamese networks), a lightweight deep learning framework that integrates evolutionary and structural embeddings to predict PPIs directly from sequence. Evaluated on multi-species benchmarks, ppIRIS achieves state-of-the-art accuracy while enabling proteome-wide screening in minutes. Trained on curated bacterial datasets and applied to the Group A Streptococcus (GAS) proteome, ppIRIS identified functional clusters associated with virulence pathways, such as nutrient transport, stress response, and metal scavenging. Extending to cross-species prediction, ppIRIS recovered 56.2% of known GAS-human plasma interactions with enrichment in complement, coagulation, and protease inhibition pathways. Experimental validation confirmed novel predictions, demonstrating the applicability of ppIRIS for systematic discovery of bacterial and cross-species PPIs. The model together with a Google Colaboratory is freely available at github.com/lupiochi/ppIRIS.
|
|
Scooped by
mhryu@live.com
Today, 1:54 AM
|
Ecosystems and human health are at serious risk due to the extensive application of pesticides in the agricultural system for controlling pests and diseases. The use of microbial consortia (MicroCons) has emerged as a promising solution for the remediation of pesticide-contaminated soil, offering a sustainable and eco-friendly alternative to physical and chemical methods; however, a systematic review on this aspect is still lacking. This comprehensive review provides an in-depth analysis of the current knowledge on microbial consortia-based remediation of pesticides in agricultural soil. Efficacy of single-strain vs multiple strains in MicroCons have been discussed to unravel the workload distribution between microbial strains in pesticide degradation. We also discuss the design and optimization of microbial consortia for remediation, highlighting the role of advanced tools and the mechanisms of MicroCons action. Furthermore, emerging trends and future directions in the field, including the potential of synthetic biology, machine learning (ML), and artificial intelligence (AI) are also covered. This review aims to critically expand the mechanistic understanding of how microbe-mediated remediation strategies might reduce pesticide phytotoxicity, enhance crop production in pesticide-stressed soils, and inspire future research and practices in MicroCons-based remediation to achieve the Sustainable Development Goals (SDGs).
|
|
Scooped by
mhryu@live.com
Today, 1:48 AM
|
Targeted protein degradation is a promising strategy for drug discovery, but designing effective PROTACs remains challenging, especially for proteins without well-defined binding sites. Current methods rely on modifying linkers between fixed ligands, which limits the diversity and innovation of the overall molecular architecture of PROTAC. Here, we introduce DeepDegradome, an AI-powered method that automates the structure-aware design of both small-molecule ligands and PROTACs. It employs a large fragment library constructed from public databases and applies an in-house docking method (iFitDock) to obtain initial binding fragments. DeepDegradome builds ligands by assembling these fragments based on the shape and physicochemical features of the target protein pocket. It can further construct PROTACs from these generated ligands, eliminating the dependency on predefined warheads or E3 ligands. Compared to other AI models, DeepDegradome produces more valid, drug-like molecules with higher predicted binding affinity. We demonstrate DeepDegradome’s effectiveness by designing and validating multiple potency inhibitors and PROTACs for two protein targets: WDR5 and CDK9. One synthesized compound showed excellent agreement between predicted and actual binding conformation confirmed by X-ray crystallography. By combining ligand and PROTAC design in one system, DeepDegradome offers a scalable and reliable tool for discovering new drugs against protein targets.
|