 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:25 AM
|
The healthy human vaginal microbiota is typically dominated by one species of Lactobacillus: L. iners, L. crispatus, L. jensenii, or L. gasseri. L. iners, the most prevalent vaginal microbe globally, is the most fastidious of the vaginal lactobacilli, has the smallest genome, and produces less lactic acid (only the L-isoform). L. iners is also less protective against bacterial vaginosis, and uniquely encodes a cholesterol-dependent cytolysin, inerolysin, suggesting it may be a pathobiont. Despite its central role in the health of over one billion females, L. iners biology remains poorly understood in part due to a lack of genetic editing tools. Here, we present findings that L. iners is naturally competent and can be transformed easily by exogenous DNA. Natural competence was leveraged to disrupt the iny gene encoding inerolysin, and comGA, encoding the ATPase component of the competence pilus. Both gene disruptions were accomplished using PCR assembled DNA fragments comprising a drug resistance gene cassette (tetM or ermB) flanked by ~2 kb regions of homology to the L. iners chromosome. We further demonstrate that comGA is essential for L. iners transformation. The ability to rapidly perform targeted deletions in L. iners with in vitro generated DNA templates provides a straightforward and much needed method to probe the genetics and physiology of these important vaginal bacteria.
|
|
Scooped by
mhryu@live.com
Today, 12:19 AM
|
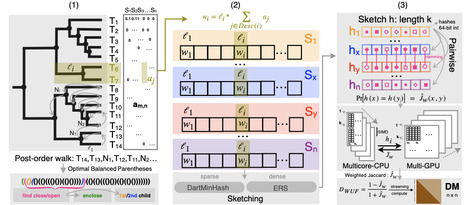
We introduce a new algorithm, DartUniFrac, and a near-optimal implementation with GPU acceleration, up to three orders of magnitude faster than the state of the art and scaling to millions of samples (pairwise) and billions of taxa. DartUniFrac connects UniFrac with weighted Jaccard similarity and exploits sketching algorithms for fast computation. We benchmark DartUniFrac against exact UniFrac implementations, demonstrating that DartUniFrac is statistically indistinguishable from them on real-world microbiome and metagenomic datasets.
|
|
Scooped by
mhryu@live.com
March 3, 11:47 PM
|
Genome architecture reorganizes over evolutionary time to support complex multicellularity without a proportional expansion of coding DNA. We conducted a cross-kingdom comparative analysis using high-quality RefSeq assemblies annotated by the NCBI Genome Annotation Pipeline, restricting the dataset to chromosome-level or complete genomes. Scaling relationships among genome size, gene content, and coding DNA content reveal compositional transitions that distinguish prokaryotic, unicellular eukaryotic, and multicellular lineages. Beyond ∼40 Mb of genic content, coding expansion slows and saturates, indicating compositional constraints that shaped the rise of multicellularity. These results establish scaling laws that quantify how noncoding sequence expansion dominates genome growth in complex eukaryotes.
|
|
Scooped by
mhryu@live.com
March 3, 11:01 PM
|
The intensifying climate crisis necessitates a global transition from fossil fuels to renewable energy sources. To meet this demand, metabolic engineering has become a pivotal strategy for developing microorganisms as efficient cell factories capable of producing fuels and fuel precursors. Among the biofuel platforms, fatty acid–based fuels are particularly promising, offering energy densities comparable to those of petroleum-based fuels. Recent advances in systems metabolic engineering, including metabolic pathway optimization, cofactor balancing, and dynamic regulation, have significantly improved the microbial production of key fuels and intermediates such as alka(e)nes, and fatty acid esters. In this review, we discuss recent progress in metabolic engineering strategies for microbial production of representative fatty acid-based fuels, highlighting current technological challenges and future directions.
|
|
Scooped by
mhryu@live.com
March 3, 10:43 PM
|
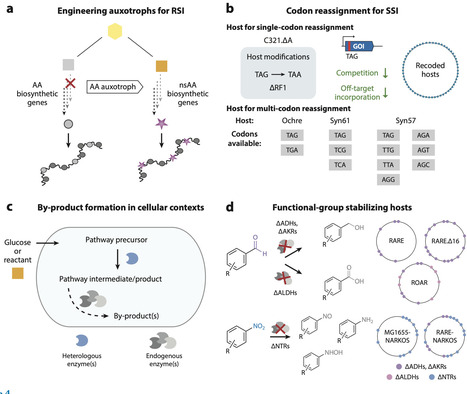
Genetic code expansion (GCE) is the ability to encode polypeptide building blocks beyond the standard 20 the ribosome uses for protein translation, known as nonstandard amino acids (nsAAs). The broadening of chemical functionalities in proteins produced by live cells has generated substantial value across fundamental and applied research settings. However, a common limitation of GCE approaches is their reliance on the supplementation of chemically synthesized nsAAs to cell culture media. To overcome this limitation of nsAA sourcing, efforts have engineered systems for nsAA biosynthesis, often in the same host that performs GCE. In recent years, these works have reported new chemical targets obtained through biosynthesis, as well as additional rationale for combining metabolic engineering and GCE, particularly for synthetic biology applications. Here, we review this rapidly advancing field and provide our perspectives on technical and conceptual innovations.
|
|
Scooped by
mhryu@live.com
March 3, 7:01 PM
|
Trimethoprim is a clinically important antibiotic used for the routine treatment of urinary tract infections as a cost-effective first-line choice for treatment. A unique feature of the drug is that it can have bacteriostatic or bactericidal effects depending upon the metabolites available in the environment. Bacteriostatic activity requires the absence of nucleoside from the growth media. Conversely, bactericidal activity requires the presence of a nucleoside and the amino acids glycine and methionine. Mechanistically, bacteriostatic action does not appear to be dependent upon protein or RNA synthesis, whereas protein synthesis does not appear to be essential for bactericidal activity. Instead, this is likely due to the inhibition of DNA synthesis and the triggering of programmed cell death, involving the suicide module mazEF. In summary, trimethoprim has a complex mechanism of action that should be considered when researching the antibiotic and informing growth media design to test susceptibility.
|
|
Scooped by
mhryu@live.com
March 3, 4:17 PM
|
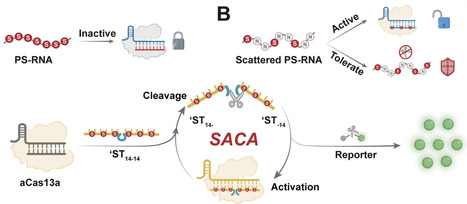
The CRISPR/Cas system is a powerful tool for molecular diagnostics, but its reliance on linear amplification constrains sensitivity, particularly for in situ imaging. Here, we discovered that phosphorothioate (PS)-modified activators can modulate Cas enzyme conformation via hydrophobic anchoring. By adjusting the PS modification sites, we achieved precise control over Cas activation and trans-cleavage resistance. Guided by this mechanism, we proposed a tailored design strategy featuring a “scattered” PS modification to engineer a linear “Coordinator” probe. This design effectively decouples Cas enzyme activation from substrate trans-cleavage resistance, enabling the construction of a Scattered PS Nucleic Acid-driven Cas Autocatalytic system (SACA). SACA achieves exponential amplification without external enzymes, enhancing Cas12a and Cas13a sensitivity by 50 000-fold and 10 000-fold, respectively. Furthermore, the superior biostability and structural simplicity of these linear probes endow SACA with excellent compatibility, facilitating precise in situ imaging of HPV16 and HPV18 mRNA in cervical cancer cells. This study not only advances the understanding of Cas enzyme regulation by chemically modified nucleic acids but also establishes a new paradigm for precise and efficient molecular diagnostics.
|
|
Scooped by
mhryu@live.com
March 3, 3:40 PM
|
Metagenomic Hi-C (metaHi-C) links mobile genetic elements to their cellular hosts directly within complex microbial communities. Once shotgun and Hi-C libraries have been generated, however, the main challenges shift to the bioinformatics required for preprocessing, genome binning, taxonomic annotation, and network-level interpretation. Here, we present metaHi-C protocols that span from raw reads to downstream data analyses. Basic Protocol 1 describes quality control of shotgun and Hi-C reads, metagenomic assembly, Hi-C read mapping, and viral contig identification from assembled contigs. Basic Protocol 2 details the use of ViralCC to recover viral metagenome-assembled genomes (vMAGs) and infer virus–host linkages. Support Protocol 1 introduces NormCC and ImputeCC for normalization of raw Hi-C contacts and host genome binning. Support Protocols 2 and 3 describe taxonomic annotation of host MAGs with GTDB-Tk and viral bins with Virgo, respectively. Support Protocol 4 shows how to integrate these outputs in MetaHiCNet to generate cross-taxa and cross-bin Hi-C interaction networks. Together, these protocols provide a reproducible workflow for reconstructing viral and host genomes, assigning consistent taxonomies, and visualizing metaHi-C–derived virus–host interaction structure across diverse microbiomes. methods
|
|
Scooped by
mhryu@live.com
March 3, 2:56 PM
|
Toxin-antitoxin-chaperone (TAC) systems are three-part gene clusters encoding a toxin, antitoxin, and specialized SecB-like chaperone (SecBTA) with emerging roles in phage defense. To identify and classify SecB homologs and associated TACs across bacteria, we surveyed the full RefSeq database. Phylogenetic and gene neighborhood analyses reveal three major SecB subfamilies: two housekeeping groups and a diverse SecBTA clade associated with eight TAC classes, five of which were previously unknown. Despite broad sequence divergence, structural predictions show conserved SecB tetrameric folds and toxin-antitoxin interfaces. The SecB chaperone phylogeny is incongruent with the identity of the TA component, suggesting modular shuffling during TAC evolution. We demonstrate toxicity of class 2 ART toxins from E. coli, Bacillus subtilis, and Streptococcus gordonii, all of which we show inhibit protein synthesis. All TAC classes can be prophage encoded, indicative of phage-driven mobility and rapid diversification.
|
|
Scooped by
mhryu@live.com
March 3, 1:59 PM
|
Trichoderma fungi support sustainable agriculture by suppressing plant diseases and improving crop performance. However, emerging pathogenicity of Trichoderma warrants further ecological and genetic characterization. Here we used machine learning to correlate genomic data from 37 Trichoderma strains with over 140 phenotypic traits, spanning metabolic versatility, biotic interactions, stress tolerance and reproductive strategies. We determined Trichoderma to be an ancient, genetically cohesive and physiologically diverse genus with spores capable of germination in water and dispersal via air and water droplets. Metabolic preferences indicate universal adaptation to mycoparasitism and to niches like arboreal microbial mats, alongside broader saprotrophic versatility. Our analyses are consistent with character displacement among close relatives and convergent evolution in distant lineages, with both processes shaping ecological plasticity and traits including dispersal modes, terrestrialization or endophytism. Our findings reveal that while some Trichoderma species show traits of biosafety concern, its vast ecophysiological diversity enables the development of safe, targeted bioeffectors. Analysis of 37 genomes together with more than 140 phenotypic traits links genomic features to ecological fitness and lifestyle diversity in Trichoderma fungi.
|
|
Scooped by
mhryu@live.com
March 3, 9:33 AM
|
Understanding how to engineer transcriptional regulation in plants is key to advancing both fundamental knowledge and practical applications in plant biology. Native gene promoters, while widely used, are constrained by evolutionary pressures that limit their modularity, tunability, and predictability across genetic backgrounds and species. Synthetic promoters, artificial DNA sequences composed of defined cis-regulatory elements (CREs) for recruitment of gene-specific transcription factors (TFs) and general transcriptional machinery, provide a powerful alternative for achieving fine-tuned transcriptional control. This review examines the design and application of synthetic promoters in plants, emphasizing current strategies, ongoing challenges, and avenues for innovation. We cover the structure of plant promoter architecture, including the contributions of core, proximal, and distal regions, and highlight how promoter grammar (i.e., motif identity, motif distance from transcription start site, spacing between motifs, helical phase of TF binding, motif orientation, and combinatorial interactions between motifs) impacts transcriptional activity. We outline how synthetic promoters are designed and validated via high-throughput reporter assays. Applications of synthetic promoters are discussed across functional genomics studies, biosensor creation, logic gate-based genetic circuits, and practical crop engineering, with examples covering constitutively expressing, hormone-responsive, pathogen-inducible, and abiotic stress-responsive promoter designs. We discuss traditional and emerging computational frameworks that enable CRE identification, novel synthetic promoter generation, and prediction of promoter sequence activity in silico to inform the rational design of promoters with predictable performance and spatiotemporal expression. We emphasize the importance of integrating experimental studies and computational approaches through iterative Design-Build-Test-Learn (DBTL) cycles to standardize and optimize frameworks for synthetic promoter development. By combining insights from plant promoter studies with advances in both plant-specific and non-plant synthetic promoter generation and computational modeling, researchers can expand synthetic promoter libraries to enable complex man-driven transcriptional regulation across various plant systems.
|
|
Scooped by
mhryu@live.com
March 3, 1:56 AM
|
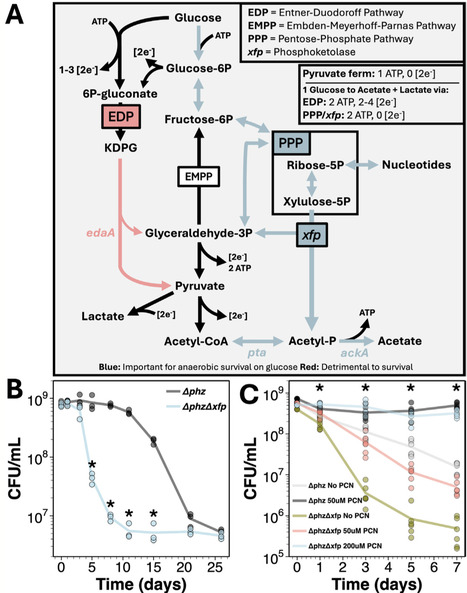
Across diverse contexts, bacteria experience loss of electron acceptors due to fluctuating environmental conditions, leading to growth-arrest and reductive stress. Yet, microbial metabolism has been primarily studied with cells growing under nutrient-replete conditions. To study how cells preserve metabolic flux under reductively stressed growth-arrest, we explored how the opportunistic pathogen Pseudomonas aeruginosa remodels its metabolism under such conditions. During anaerobic survival on glucose, P. aeruginosa utilizes the upper Embden-Meyerhoff-Parnas pathway and pentose-phosphate pathway to generate metabolite precursors for a previously undescribed phosphoketolase (herein termed xfp) used to produce acetyl-P and indirectly ATP via subsequent acetate formation. This re-routing bypasses P. aeruginosa's canonical glucose-catabolizing Entner-Doudoroff pathway (EDP), allowing for metabolic flux without exacerbating reductive stress. Moreover, anaerobic survival on diverse carbon sources triggers purine degradation and metabolite accumulation, requiring xfp to maintain metabolic balance and viability. Thus, our data suggest that phosphoketolases may play an additional role in ribonucleotide balance. This study expands our understanding of P. aeruginosa's anaerobic survival strategies and serves as a reminder that large gaps remain in our understanding of growth arrest physiology even in well-studied model organisms, highlighting the potential for basic discovery in the realm of non-growth metabolism.
|
|
Scooped by
mhryu@live.com
March 3, 12:40 AM
|
While protein language models (PLMs) have shown great promise for protein design, their performance is fundamentally constrained by the diversity and completeness of available training data. In particular, PLMs often struggle to extrapolate to sequences that fall outside the distribution spanned by their training sets, limiting their ability to discover proteins in sparsely sampled regions of sequence space. Here we test the hypothesis that experimentally expanding training diversity can convert extrapolation into interpolation and thereby enable discovery of functional sequences beyond natural protein manifolds. Using large-scale gene synthesis and DNA shuffling, we generate libraries that span a broad region of fluorescent protein sequence space and create chimeric variants that bridge between distant homologs. Functional screening for blue fluorescence yields thousands of active variants distributed across diverse sequence lineages. Fine-tuning ProtGPT2 on this expanded dataset enables generation of diverse fluorescent proteins, including designs that extend beyond the regions occupied by known natural sequences while retaining function. This work illustrates how synthetic approaches can help address key limitations in machine learning-guided protein design, especially for small or sparsely populated protein families, by actively creating novel sequences across unexplored but functional regions of sequence space.
|
|
|
Scooped by
mhryu@live.com
Today, 12:21 AM
|
Marine plastic litter, including microplastics, has a profound impact on the ocean and its wildlife, and strategies to remove/eliminate it are needed. Microbial biodegradation, particularly by bacteria, offers a potential solution, where a link between hydrocarbon and plastic-degradation has been hypothesized. This study screened the plastic-degrading potential of 18 bacterial strains isolated from 1-month-old biofilms developed in three submerged plastic fishing nets (braided polyethylene (PE), braided nylon, thin nylon). In addition, three highly efficient hydrocarbon-degrading strains were also tested. Strains were cultivated on solid minimal media with fishing net small pieces (new/unused nets) added as a carbon source for 1 month, followed by tributyrin-agar assays to assess esterase/lipase activity. Eleven bacteria exhibited enhanced growth with net polymers, mainly from the genera Sulfitobacter, Rhodococcus, Bacillus, and Pseudomonas, and eight of which bacteria also demonstrated esterase/lipase activity. Then, genes encoding hydrocarbon or plastic-degrading enzymes (alkB and almA homologs, PETase-like enzymes) were screened by PCR in the 21 mentioned bacteria and in ca.100 other strains found in submerged nets biofilm. Amplification of the investigated genes was predominantly observed in Actinomycetes strains. Genome mining of six promising strains revealed hits with enzymes linked to degradation of synthetic polymers like polyethylene terephthalate, low-density PE and nylon. The workflow developed here enabled the selection of marine bacteria with plastic-degrading potential, sourced from biofilms of submerged plastic fishing nets and hydrocarbon-enriched environments.
|
|
Scooped by
mhryu@live.com
Today, 12:17 AM
|
From soil to the gut, communities composed of thousands of microbes perform functions such as carbon sequestration and immune system regulation. Here, we introduce a data-driven approach that explains how community function can be traced to just a few groups of microbes or genes. In gut communities, our neural-network based clustering algorithm correctly recovers known functional groups. In the ocean metagenome, it distills ~500 gene modules down to three sparse groups highlighting survival strategies at different depths. In soils, it distills ~4400 bacterial species into two groups that enter a mathematical model of nitrate metabolism. By combining interpretable ML with strain isolation and sequencing experiments, we connect the metabolic specialization of each group to community-wide responses to perturbations. This integrated approach yields simple structure-function maps of microbiomes, allowing the discovery of molecular mechanisms underlying human and environmental health. More broadly, we illustrate how to do function-informed dimensionality reduction in biology.
|
|
Scooped by
mhryu@live.com
March 3, 11:29 PM
|
Natural evolution is high-dimensional; organisms adapt to many pressures at once, across substrates, environments, and genetic backgrounds. Yet most directed evolution methods flatten this landscape to a single selection axis, hiding tradeoffs, and limiting what can be learned. Phage-assisted continuous evolution (PACE) is uniquely suited for multivariate selection because horizontal gene transfer couples genotype to propagation and allows the same phage lineage to traverse different selection environments. In practice, implementing this at scale has been prohibitive because each selection demands its own host culture, and every culture must be held for days to weeks within a narrow, infectable density window using continuously responsive bioreactors. In this work, TurboPRANCE is presented as an open-source, queueable robotic platform that integrates ~200 independently controlled turbidostats with 96 parallel PACE lagoons under closed-loop control. Each turbidostat operates as a fully separate unit that can be equilibrated and initiated on its own schedule, enabling asynchronous starts and sustained operation without intervention. Automated media formulation, programmable dosing, on-deck sterilization, and adaptive scheduling coordinate growth control with the changing needs of the robotic workflow, dynamically adjusting dilution and transfer timing around formulation, sampling, and handling steps to keep each culture at consistent infectable densities despite unpredictable method demands. Cultures can be multiplexed and titrated into lagoons at defined ratios, swapped in and out on a schedule, or kept fully separate across experiments, creating a combinatorial space of selection pressures and programs that is effectively unbounded. Additionally, to enable high-throughput evolutionary tracking that scales with TurboPRANCE, Nanopore long-read sequencing was combined with DeepVariant, a deep learning-based variant caller, enabling population-level tracking of evolving variants. The result is a system that generates high-resolution time-resolvable evolutionary trajectories and large parallel datasets spanning diverse selection regimes, yielding dense, multivariate training data to map and engineer complex fitness landscapes at scale.
|
|
Scooped by
mhryu@live.com
March 3, 10:53 PM
|
Traditional 16S rRNA gene and Internal Transcribed Spacer region amplicon sequencing provides only relative abundance, often leading to biased ecological interpretations. To overcome this limitation, we developed Accu16S/AccuITS, an absolute quantification method for bacterial and fungal amplicons based on synthetic internal spike-in DNA with known copy numbers. By adding internal standards prior to PCR and sequencing, absolute microbial abundances can be calculated using standard curve regression. Accu16S/AccuITS exhibits sensitivity and consistency comparable to quantitative PCR and is applicable to diverse sample types. A single sequencing run simultaneously yields relative abundance, total absolute abundance, and taxon-specific absolute abundance. Case studies across diverse ecosystems demonstrate that absolute quantification provides ecologically and functionally meaningful insights beyond those obtained from relative abundance analyses.
|
|
Scooped by
mhryu@live.com
March 3, 10:39 PM
|
Filamentous fungi, particularly Aspergillus species, play a crucial role in industrial biotechnology due to their exceptional protein secretion systems and robust metabolic adaptability. However, variability in heterologous protein production presents a major bottleneck. Here, we combine multi-omics analyses and synthetic biology to identify msnA as a key regulator of recombinant protein production in Aspergillus spp. Overexpression of msnA enhances secretion of diverse proteins (e.g., glycoside hydrolases, fluorescent proteins) by up to 2.8-fold in A. nidulans, A. niger, and A. oryzae, demonstrating functional conservation across Aspergillus species. Mechanistically, transcriptomic studies reveal that msnA reprograms cellular resource allocation by upregulating vesicle transport and fatty-acid degradation while downregulating secondary metabolism, thereby redirecting energy toward recombinant protein synthesis and secretion. Remarkably, the overexpression of msnA significantly improves the efficiency of recombinant protein secretion, even with lower transcriptional expression. In some species, this enhancement occurs despite a decrease in total secreted protein. Our research identifies msnA as a potential target gene for optimizing fungal cell factories, offering a framework to enhance recombinant protein production and increase the efficiency of Aspergillus-based systems in industrial applications.
|
|
Scooped by
mhryu@live.com
March 3, 6:48 PM
|
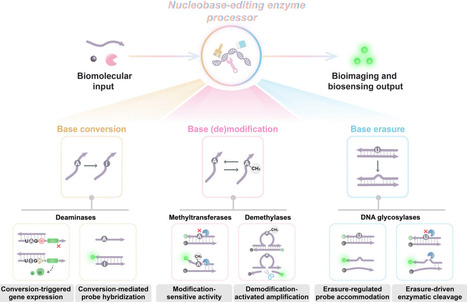
Nucleobases, the fundamental units of DNA and RNA, undergo diverse chemical modifications and lesions that regulate genomic stability, epigenetic states, and cellular functions. Nucleobase-editing enzymes, including deaminases, methyltransferases/demethylases, and DNA glycosylases, catalyze precise base conversions, modification/demodification, or excision reactions at high resolution without inducing double-stranded breaks. Originally studied in transcriptomic diversification, epigenetic regulation, and DNA repair, these enzymes are now increasingly repurposed as programmable actuators in biosensing and bioimaging applications. In this review, we first outline the catalytic principles of representative nucleobase-editing enzymes, emphasizing substrate recognition, reaction mechanisms, and physiological functions. We then highlight how these enzymes are integrated into biosensing and bioimaging modules across three major modes: nucleobase conversion, where site-directed deamination enables facile fluorescent protein reporter translation or specific DNA probe hybridization signals; nucleobase modification/demodification, where methylation and demethylation events regulate downstream selective enzymatic biocatalysis or programmble functional nucleic acid activation; and nucleobase erasure, where glycosylase-mediated base excisions facilitate specific probe accommodation or effient enzyme-catalyzed amplification. Such nucleobase-editing enzyme-driven systems offer high specificity, efficient amplification, and high compatibility with physiological conditions, enabling sensitive and spatiotemporally resolved monitoring of nucleic acids, proteins, and cellular processes. Finally, we discuss the advantages, challenges, and future directions of this emerging field. Advances in enzyme engineering, delivery strategies, and multiple circuitry integration are expected to yield next-generation bioanalytical platforms with improved precision, scalability, and clinical applicability. Collectively, these developments establish nucleobase-editing enzymes as a versatile molecular toolbox that bridges fundamental enzymology with applied biotechnology for diagnostics, therapeutic monitoring, and synthetic biology.
|
|
Scooped by
mhryu@live.com
March 3, 4:13 PM
|
One-pot CRISPR-based diagnostics have transformed nucleic acid testing, yet their design customizability remains constrained. Because target programming and cis-cleavage activity are simultaneously determined during CRISPR RNA (crRNA) design, optimizing cleavage activity to match isothermal amplification inevitably requires altering the programmed crRNA sequence. This requirement fundamentally constrains the range of compatible target sequences, imposing limitations on the flexible design of diagnostic assays. Here, we establish a customizable one-pot system by decoupling the dual functions inherent in crRNA design to enable their independent control. In this strategy, target programming remains defined by the crRNA sequence, whereas cis-cleavage activity is regulated by the reaction energy barrier. We selectively modulate this energy barrier through the introduction of a crRNA–complementary RNA oligonucleotide, achieving cleavage regulation without altering the crRNA sequence. Consequently, this approach ensures that cis-cleavage activity matches isothermal amplification conditions independent of the programmed target sequence, thereby realizing a customizable CRISPR diagnostic system. We validated the clinical applicability of this system using 120 patient-derived samples, achieving sensitivity and specificity comparable to quantitative polymerase chain reaction. Collectively, this work resolves a fundamental constraint of CRISPR diagnostics and establishes a customizable and clinically deployable platform for next-generation nucleic acid testing.
|
|
Scooped by
mhryu@live.com
March 3, 3:04 PM
|
Polyhydroxyalkanoates (PHA) are promising biodegradable alternatives to petroleum-based plastics, but high production costs limit their deployment. In this study, we evaluated a cosubstrate strategy combining vanillic acid (VA), representative of lignin-derived aromatics, with palm oil (PO) recovered from oil palm fruit residues to enhance PHA synthesis in Pseudomonas putida. Cofeeding 4 g/L VA and 4 g/L PO increased PHA titer to 1.13 g/L, a 52.7% improvement over VA alone, and yielded a new monomer, 3-hydroxyhexanoate (3HHx). Integrated transcriptome and metabolomic analyses revealed that co-substrate metabolism attenuated TCA flux, enhanced β-oxidation and hydroxyacyl precursor pools, and altered NAD(P)H turnover and membrane lipids. These shifts were correlated with expanded monomer diversity. This study demonstrates that cofeeding lignin-derived aromatics with agricultural oils provides a synergistic route to improve PHA yield and diversify monomer composition, linking biomass valorization with circular, low-carbon material production.
|
|
Scooped by
mhryu@live.com
March 3, 2:45 PM
|
The frequent occurrence of harmful algal blooms (HAB) poses severe threats to aquatic ecosystems, aquaculture industries and human health. Recently, algicidal bacteria have emerged as a promising biocontrol strategy. However, the precise mechanisms underlying their algicidal effects remain poorly understood, limiting their practical application in environmental management. This review systematically summarises the interactions between bacteria and algae, as well as the various algicidal modes employed by bacteria, with a particular focus on the mechanisms driving bacterial algicidal activity. Key bacterial behaviors such as chemotaxis, adhesion, quorum sensing and the release of extracellular vesicles have been identified as critical factors in the algicidal process, among which the role of bacterial extracellular vesicles warrants special attention. Furthermore, we elaborate on the death mechanisms of algal cells upon bacterial attack, including loss of cellular structural integrity, impairment of photosynthetic systems, oxidative stress responses and disruption of calcium ion homeostasis. Notably, advancements in detection technologies have increasingly highlighted the importance of calcium signalling regulation in algal cell death. This review not only elucidates the molecular mechanisms of bacterial algicidal activity, providing a theoretical foundation for the biocontrol of red tides, but also deepens our understanding of bacteria-algae interactions.
|
|
Scooped by
mhryu@live.com
March 3, 9:43 AM
|
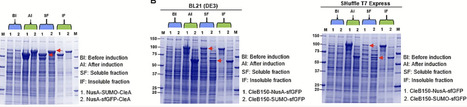
The Clostridium estertheticum complex (CEC) consists of closely related bacterial species that are mostly isolated from meat processing environment. Genome mining studies have recently established CEC as a source of class I bacteriocins. However, up until now, class II bacteriocins have not been reported from CEC. In the present study, we determined the presence of class II bacteriocin biosynthetic clusters in 33 CEC genomes through genome mining followed by bacteriocin production through heterologous expression in E. coli. Six biosynthetic gene clusters belonging to class IIa (n = 1), IIb (n = 2), IId (n = 2) and an undefined class containing three precursor peptides were identified in six different CEC strains. Using molecular biology, we developed dedicated expression vectors for the class IIa bacteriocin, clesteriocin A. Its precursor peptide, CleA, and protease, CleB150, were initially expressed in insoluble form in E. coli. Through a systematic analysis of suitable solubility enhancing protein tags, both CleA and CleB150 were expressed in significant amounts of soluble fractions using tandem tags derived from Small Ubiquitin-like Modifier, superfolder Green fluorescent protein, Maltose binding protein and N-utilization substance. Mature clesteriocin A was obtained following in vitro maturation reactions between the tagged CleA and CleB150. The novel bacteriocin displayed antimicrobial activity against Listeria monocytogenes, which was mediated by the mptC gene. By combining genome mining and molecular biology, we have shown CEC is a source of novel class II bacteriocins that have potential for application as food biopreservatives.
|
|
Scooped by
mhryu@live.com
March 3, 2:01 AM
|
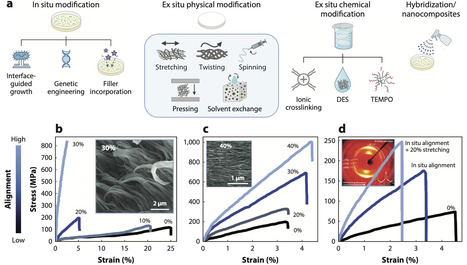
Polymer design is being reshaped by demands for low-carbon fabrication and bioactive/living function. We trace the bidirectional interface between microbes and polymers. First, we analyze how microbes synthesize polymers like polysaccharides, polyesters, and proteins and how post-synthesis processing (via mechanical and/or chemical treatments) reshapes molecular architecture and mechanical and thermal properties. We compare reported properties, highlight missing metrics, and evaluate sustainability levers including solvent recovery, cradle-to-gate impacts, and biodegradation/biocontainment constraints. Second, we examine how polymers shape the behavior of living organisms in the context of engineered living materials. Design is organized around four axes—regulating adhesion and detachment, sustaining or directing growth for regeneration, imposing spatial organization on consortia, and tuning phenotype—with implementations in drug delivery, carbon capture, antimicrobial screening, and structural composites. Finally, we outline how automation, artificial intelligence–guided experimentation, and robust sustainability metrics can couple performance with responsible deployment.
|
|
Scooped by
mhryu@live.com
March 3, 1:45 AM
|
A long-standing goal in biomedical research is to label and manipulate intracellular targets, which could be achieved through the cytosolic delivery of exogenous functional proteins. The development of Tat clusters has advanced the nontoxic intracellular delivery of functional antibodies at low concentrations, but the variety of proteins that can be successfully delivered remains limited. Here, we find that by simply reversibly modifying the surface of functional proteins with anionic peptide patches, various protein cargoes (which are normally difficult to deliver) can be delivered into living cells by synergetic electrostatic interactions with the cationic cell-penetrating peptide clusters TAT3. To demonstrate the applicability of this approach, we successfully deliver functional proteins with widely varying molecular weights (∼1.5 kDa to 430 kDa) and isoelectric points (less than 5 to greater than 9) into the cytosol of cells. By exploiting this method, we also achieve protein delivery in plant tissues, which is more challenging due to the presence of intact plant cell walls. This strategy is further applied for the cytosolic delivery of synthetic protein probes carrying posttranslational modifications (PTMs), which can aid in in situ mapping of the intracellular PTM-mediated interactome. Overall, this strategy is expected to enrich cytosolic protein delivery technology and help to repurpose a wide range of customized and therapeutic proteins for emerging intracellular applications. The development of Tat clusters has advanced the intracellular delivery of functional antibodies at low concentrations, but the variety of proteins that can be successfully delivered remains limited. Here, the authors report that by reversibly modifying the surface of functional proteins with anionic peptides, various protein cargoes (otherwise difficult to deliver) can be delivered into living cells by synergetic electrostatic interactions with the cationic cell-penetrating peptide clusters TAT3.
|




























1str