 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:00 PM
|
Large language models (LLMs) are increasingly used by plant biologists to summarize literature, generate hypotheses, and interpret experimental results. However, LLMs are unreliable sources of exhaustive, source-attributed facts, a critical limitation for the list-style queries that pervade plant biology (e.g., "list all transcription factors regulating secondary cell wall (SCW) biosynthesis in Arabidopsis"). Here, we query ChatGPT, Claude, and Gemini with such queries and demonstrate that none return complete gene lists with reliable citations. We trace these failures to how LLMs store knowledge: as statistical patterns distributed across billions of internal parameters, with no mechanism to guarantee completeness, provenance, or reproducibility. We also review fine-tuning mitigation strategies, including multi-task instruction tuning, parameter-efficient methods, and context engineering, that alleviate but do not resolve these limitations. We then discuss retrieval-augmented generation (RAG), which feeds relevant documents to the LLM at query time, and argue that while it improves source attribution, it remains impractical when answers require synthesizing information scattered across hundreds of papers. As an alternative, we advocate graph retrieval-augmented generation (GraphRAG), in which the LLM serves as a reasoning and language interface over a structured, provenance-linked knowledge graph (KG) that returns complete result sets reproducibly. We outline a practical GraphRAG architecture and survey existing plant KG resources. Finally, we discuss open challenges, including entity disambiguation, relation normalization and evidence grading, and propose a roadmap for building open, continuously updated plant KGs that can turn "read 1,000 papers" into a single reproducible query.
|
|
Scooped by
mhryu@live.com
Today, 10:31 PM
|
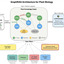
The auxin-inducible degron (AID) system enables targeted protein degradation in vivo. Conventionally, the AID tag is fused directly to the terminus of the target protein; however, its intrinsically disordered nature and destabilizing motifs can render it susceptible to premature proteolytic removal from its tagged protein, resulting in incomplete degradation upon auxin induction. In addition, direct terminal fusion of the conventional AID tag may compromise protein stability. To address these limitations, we introduce a new degron design in which the conventional AID tag, mIAA7, is inserted into an exposed loop of green fluorescent protein (GFP). The resulting engineered GFP variant, termed the “constrained AID” (cAID) tag, was validated in the industrially important oleaginous yeast Yarrowia lipolytica. Inserting mIAA7 into GFP, rather than fusing it directly at the GFP terminus, elevated expression of the GFP fusion protein and enabled more complete degradation upon auxin induction. The utility of the novel cAID tag was further validated by tagging a soluble cytosolic anti-mCherry nanobody and by targeting β-carotene ketolase, squalene synthase, and squalene epoxidase to modulate carotenoid biosynthesis. Compared to direct fusion with the conventional AID tag, cAID tag enabled more complete protein degradation and tighter temporal control of metabolism, while minimizing perturbation to the tagged protein.
|
|
Scooped by
mhryu@live.com
Today, 4:41 PM
|
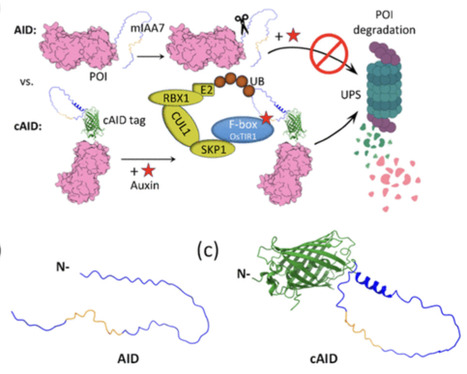
Bacterial secreted proteins, particularly effectors delivered by specialized secretion systems, are key mediators of virulence and host–pathogen interactions. However, accurate computational identification remains challenging, as many existing methods rely heavily on sequence similarity or handcrafted features, and often focus on a single secretion system. Recent studies have reported that some bacterial effectors may be associated with more than one secretion system, highlighting the complexity of secretion system annotation and motivating the development of system-aware computational prediction approaches. Here, we present PLM-Effector, a hybrid deep learning framework that integrates modern protein language models (PLMs) with multiple neural architectures via a two-layer ensemble stacking strategy. By extracting complementary features from N- and C-terminal regions, PLM-Effector enables secretion-type-aware prediction across five major bacterial secretion systems (T1SS–T4SS and T6SS), with each system modeled independently. Systematic benchmarking shows that embeddings from protein-specific PLMs (ESM-1b, ESM2_t33, ProtT5) are more discriminative than those from general-purpose language models (e.g. BERT, BioBERT). Leveraging these representations, PLM-Effector achieves superior performance on an independent test set, with macro F1-scores of 0.9848, 0.8649, 0.9899, 0.9620, and 0.9728 for secreted proteins of T1SS–T4SS and T6SS, respectively, outperforming existing tools and homology-based baselines. Implemented as an accessible web server (http://www.mgc.ac.cn/PLM-Effector/) with source code and datasets available (https://github.com/zhengdd0422/PLM-Effector/), PLM-Effector provides a reproducible and user-friendly platform for both small-scale and genome-wide secreted protein discovery, facilitating advances in the study of bacterial pathogenesis.
|
|
Scooped by
mhryu@live.com
Today, 4:28 PM
|
Metabolic burden arises from the reallocation of cellular resources, often resulting in stress-associated phenotypes and compromised cellular performance. However, the molecular mechanisms by which industrial microorganisms perceive and alleviate such burdens remain largely unexplored. Here, we present an online monitor to quantify metabolic burden imposed by genetic and environmental perturbations in the workhorse Corynebacterium glutamicum. RNA-seq analysis revealed a shared host response and enabled the identification of several early-responsive promoters through in vivo burden assays. Leveraging these elements, particularly the cg1940 promoter, we successfully engineered dynamic feedback systems to alleviate metabolic burden associated with suboptimal expression. Notably, the identified promoter retained burden responsiveness in E. coli, suggesting potential cross-species applicability. As a proof of concept, this feedback controller was applied to improve cell growth, protein synthesis, and chemical bioproduction. This technology offers a strategy for bolstering the robustness of C. glutamicum and potentially other microorganisms.
|
|
Scooped by
mhryu@live.com
Today, 4:12 PM
|
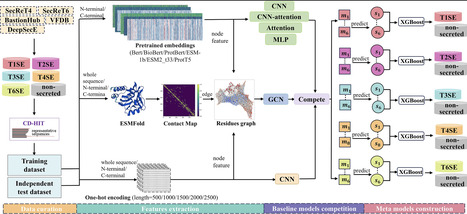
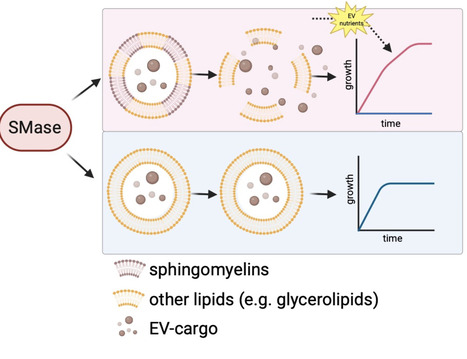
Bacterial extracellular vesicles (EVs) are known to mediate intercellular communication, virulence, and immune modulation. Here we show that bacteria can utilize EVs also as recyclable nutrient reservoirs. Using Bacillus cereus as a model organism, we demonstrate that EVs exhibit distinct dynamics depending on growth conditions: EVs produced in complex nutrient-rich media undergo time-dependent degradation, while those produced in defined nutrient-limited conditions remain stable and accumulate. We observe similar EV degradation patterns in Staphylococcus aureus. Time-resolved multi-omics profiling reveals that EVs containing the lipid sphingomyelin undergo progressive degradation. Using pharmacological inhibition, knockout mutants, and enzymatic complementation, we show that this process is driven by secreted sphingomyelinase (SMase). This enzyme contributes to degradation of sphingomyelin-containing EVs, thereby releasing their biomolecular cargo which can be used as a nutrient source. Growth assays confirm that SMase-mediated EV degradation provides a growth advantage when nutrients become depleted, thus establishing EVs as dynamic nutrient reservoirs. Bacterial extracellular vesicles can mediate intercellular communication, virulence, and immune modulation. Here the authors show that bacteria can also utilise these vesicles as recyclable nutrient reservoirs, inducing their degradation when nutrients are scarce.
|
|
Scooped by
mhryu@live.com
April 5, 12:08 PM
|
Gene drives offer revolutionary potential for the management of problematic plant populations, such as invasive weeds and herbicide-resistant species, by rapidly spreading desired genetic alterations. Two recent studies have provided experimental demonstrations of engineered CRISPR gene drive systems in plants (CAIN and ClvR). However, the successful application of such systems in the field will critically depend on an accurate understanding of plant-specific life-history traits, especially seed dormancy, a ubiquitous yet frequently overlooked eco-evolutionary force. In this study, we develop a comprehensive modelling framework for gene drives in plant populations that incorporates a persistent soil seed bank. We show how the presence of a seed bank can substantially slow gene drive spread but also reduce the genetic load required to achieve population elimination. Furthermore, we show that seed banks substantially increase the required introduction frequency of threshold-dependent gene drives, which could prevent establishment in some cases, yet also provide an intrinsic biosafety mechanism for confining a highly efficient drive to a target population. Our study highlights the need to incorporate seed-bank dynamics into gene drive strategies to ensure realistic predictions and successful field applications. The authors present a comprehensive plant-specific modelling framework for CRISPR gene drives: dormant seed banks can slow spread and require larger releases but can also ease weed elimination and limit unintended spread to nearby populations.
|
|
Scooped by
mhryu@live.com
April 5, 12:00 PM
|
Mapping protein-protein interactions (PPI) at structural resolution is essential for understanding cellular machinery. While tools like AlphaFold enable proteome-wide structural predictions, the field lacks high-throughput experimental methods to verify these predictions at scale and establish empirical thresholds for confidence metrics, such as the widely used interface predicted Template Modeling (ipTM) score. We developed LUCIA (LUminescent Cell-free Interaction Assay), a rapid biochemical screening platform bypassing traditional cloning and protein expression to validate direct binary interactions within days. Applying this to herpesviruses, clinically relevant human pathogens with large proteomes, we generated an exhaustive computational interactome of 23,215 AlphaFold-predicted dimer models across three species (HSV-1, HCMV, and KSHV), accessible via our HerpesPPIs database. Using the HSV-1 interactome as a benchmark, testing 83 high-confidence predicted dimers with LUCIA yielded 23 novel, experimentally validated interactions. By calibrating AlphaFold metrics against this direct binding data, we demonstrated that an ipTM score ≥ 0.80 identifies bona fide interactions with a positive predictive value of 77%. To demonstrate the functional power of this pipeline, we characterized a previously unknown interaction between HSV-1 UL42 and UL8, linking the viral DNA polymerase and helicase-primase complexes. Structure-guided mutations at the predicted and LUCIA-verified UL42-UL8 interface strongly reduced binding in vitro and completely abolished viral replication in cell culture. Combining the scalable LUCIA platform with computational predictions enables researchers to rapidly translate atomic-level models into validated biological mechanisms without the need to solve experimental structures.
|
|
Scooped by
mhryu@live.com
April 5, 11:19 AM
|
Transposable elements (TEs) are major drivers of genome evolution, yet their annotation and classification remain inconsistent and hard to reproduce across species. Fragmented repeats, lineage-specific innovations, and heterogeneous taxonomies across databases and tools complicate comparisons and slow progress in TE biology. To address this, we developed PanTEon, a cross-kingdom deep learning framework for reproducible TE classification that combines a harmonized database with an open, modular benchmarking platform. The PanTEon Database is an automatically curated, taxonomically broad TE repository spanning animals, plants, and fungi. The PanTEon platform standardizes training, evaluation, and inference across nine Machine Learning methods, while remaining extensible to user-defined architectures. Using this framework, we benchmark state-of-the-art Machine Learning-based TE classifiers across TE superfamilies and major eukaryotic lineages and find that performance varies markedly by kingdom and superfamily. Ensemble approaches and phylum-specific models improve predictive F1 scores, but cross-species generalization remains a major challenge. Together, PanTEon Database and PanTEon platform provide a reproducible, scalable, and extensible foundation for TE classification, enabling standardized evaluation of future AI methods and supporting community-driven annotation efforts.
|
|
Scooped by
mhryu@live.com
April 4, 5:07 PM
|
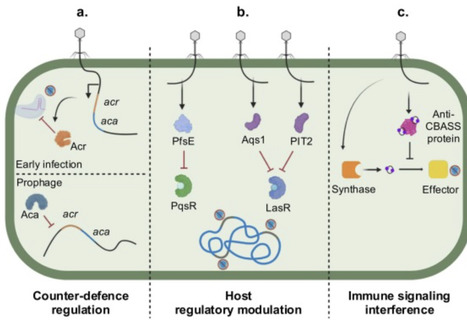
Gene regulation has emerged as an important determinant of phage infection outcomes. Host susceptibility and immunity are often governed by conditional gene expression, which allows reversible shifts in receptor availability and defence system activity that balance phage resistance with fitness costs. Phages likewise rely on tightly regulated gene expression, precisely timing counter-defence deployment and manipulating host transcriptional programmes as well as immune signalling pathways. These dynamics position gene regulation as a major determinant of bacteria-phage co-evolution, acting alongside the gain and loss of defence and counter-defence genes. Viewing phage–host interactions through gene regulation provides insight into variability in infection dynamics and helps explain why genomic information alone cannot accurately predict phage activity.
|
|
Scooped by
mhryu@live.com
April 4, 4:58 PM
|
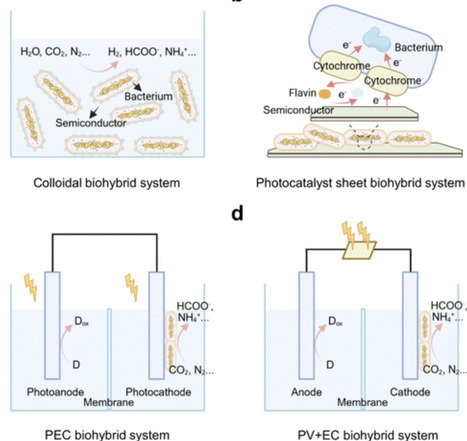
Integrating semiconductors with microorganisms is attracting significant attention as a sustainable platform for solar-to-chemical conversion. This semibiological design combines the excellent light harvesting ability of semiconductor materials with intracellular biocatalytic pathways to enable efficient solar energy conversion into complex products with high selectivity. However, the effectiveness of this interdisciplinary biohybrid approach relies on a complex interfacial biotic–abiotic interaction, and it remains challenging to construct efficient and stable microbe–semiconductor systems for practical applications. In this review, we provide a systematic overview of the fundamental mechanisms behind microbe–semiconductor systems with an emphasis on interfacial electron transfer and highlight recent advancements in the assembly of biohybrids for solar-driven biosynthesis using nonphotosynthetic bacteria. First, we provide a comprehensive introduction of semibiological photosynthesis with an emphasis on extracellular electron transfer at the biotic–abiotic interfaces. Then, we discuss the engineering of biohybrid interfaces, the characterization of microbe–semiconductor interfacial electron transfer, and their deployment in solar-to-chemical conversion. We conclude by exploring the challenges in developing and optimizing biotic–abiotic interfaces as well as providing an outlook for potential future innovations. This review therefore presents the basic principles and provides guidance for the development of semibiological photosynthetic systems.
|
|
Scooped by
mhryu@live.com
April 4, 4:32 PM
|
Long-read sequencing has shown a rapid technological development during the last years. It has been established as the standard method for the sequencing of plant genomes and has also gained importance for full plasmid sequencing. As Sanger sequencing has a limited read length of about 1 kb, long read sequencing offers a great advantage, as the full plasmid can be sequenced in one read. Here, we present a cost-effective workflow to sequence full plasmids and compare the results against an expectation. The per plasmid cost of this workflow is determined by the number of plasmids investigated simultaneously, but can be lower than the price of a single Sanger sequencing reaction. We developed a workflow for automatic data processing, which allows us to complete sequencing and data analysis within a day.
|
|
Scooped by
mhryu@live.com
April 4, 4:28 PM
|
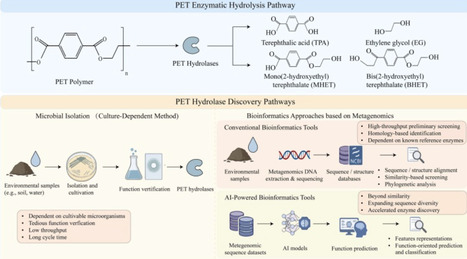
Polyethylene terephthalate (PET) hydrolases efficiently hydrolyze the ester bonds in PET, converting it into valuable monomers or oligomers, offering a sustainable biological solution to global PET plastic pollution. However, the large-scale development of high-performance PET hydrolases remains challenging due to limitations in traditional enzyme resource mining methods, including low throughput and lengthy cycles. Recent advances in artificial intelligence (AI) provide novel methodologies to overcome these challenges. This review systematically summarizes how AI empowers the high-throughput screening of PET hydrolases from mass biological databases, while allowing the accurate prediction of enzyme structures and functions. Furthermore, it critically analyzes AI-driven strategies for enzyme molecular engineering and highlights the emerging frontier of AI-assisted de novo enzyme design. By systematically evaluating the advantages and challenges of AI models in the research of PET hydrolases, this review provides an integrated technical framework and theoretical foundation to guide future innovation in enzyme mining and plastic biodegradation.
|
|
Scooped by
mhryu@live.com
April 4, 3:20 PM
|
Modern molecular analyses have revolutionized the study of microbial communities, yet DNA extraction and sequencing remain critical sources of bias. This study investigated the impact of seven different DNA extraction protocols and two 16S rRNA hypervariable regions (V1–V3 and V3–V4) on the profiling of a complex anaerobic fermentative biomass selected for medium-chain fatty acids production. Microscopic analysis established a baseline community dominated by Actinobacteria (53% ± 2%) and Firmicutes (47% ± 3%). The results demonstrate that Kit1 and Kit5 provided the highest DNA yields (up to 603 ng/μL) and the most effective recovery of these hard-to-lyse phyla, although they introduced a slight taxonomic bias toward Actinobacteria. In contrast, protocols relying on intensive chemical lysis without robust mechanical disruption (Kit4) significantly underestimated total bacterial abundance and showed the lowest purity. 16S rRNA gene sequencing revealed that the V3–V4 region provided higher alpha-diversity and a more balanced representation of the community core compared to V1–V3, which was more susceptible to extraction-related variability and overrepresented the genus Olsenella. Our multi methodological approach reveals significant biases introduced by both extraction technique and 16S rRNA gene region. This evidence highlights that protocol optimization is mandatory for achieving an accurate and comprehensive characterization of microbial ecosystems.
|
|
|
Scooped by
mhryu@live.com
Today, 10:55 PM
|
Understanding the role of gene expression in cellular function and tissue organization requires spatial and quantitative detection of individual RNA molecules. Yet, the widespread adoption of automated single-molecule fluorescence in situ hybridization (smFISH) has been limited by the cost of equipment and the complexity of experimental procedures. We present autoFISH, an affordable, user-friendly platform that removes these barriers through open-source hardware components, accessible control software, and integrated analysis tools. The system demonstrates broad applicability by enabling both conventional and signal-amplified smFISH protocols and incorporates an optimized tissue-clearing method that preserves nuclear structures. Testing across multiple cell types and tissue preparations validates the system’s reliability and reproducibility, offering a practical solution for scaling spatial transcriptomics research and advancing discoveries in cellular and developmental biology, while significantly reducing costs and the technical expertise required. AutoFISH provides a cost-effective, open-source toolbox for automated smFISH, offering integrated hardware, software, and optimized protocols to streamline spatial gene-expression profiling in cell lines and cleared tissue samples.
|
|
Scooped by
mhryu@live.com
Today, 4:54 PM
|
A systematic understanding of cellular metabolism is essential for engineering yeast and uncovering the principles of metabolic robustness and evolution, yet much of its metabolic space remains unexplored. Although yeast genome-scale metabolic models have been reconstructed and curated for over two decades, more than 90% of the yeast metabolome remains uncovered. Here, to address this gap, we have developed an integrated workflow that combines retrobiosynthesis, deep learning-based enzyme annotation and enzyme–substrate prediction to systematically explore yeast underground metabolism. Using the framework, we reconstruct a yeast metabolic twin model, Yeast-MetaTwin, comprising 16,244 metabolites, 1,976 metabolic genes and 59,865 reactions. The model reveals systematic differences in Km distributions between the known and underground networks and identifies key hub metabolites linking the underground network. Moreover, Yeast-MetaTwin predicts by-product formation in yeast cell factories, and we experimentally validate two genes converting geraniol to geranial during geraniol biosynthesis. Cellular underground metabolism plays crucial roles in enzyme promiscuity, metabolism and biological evolution, but it has hardly been investigated. Here the authors combine retrobiosynthesis with deep learning enzyme annotation and enzyme–substrate prediction methods to explore it, reconstructing the yeast metabolic twin model, Yeast-MetaTwin.
|
|
Scooped by
mhryu@live.com
Today, 4:29 PM
|
Bioart and transgenic art employ living and genetically engineered organisms. This article examines how European biosafety governance influences the public presentation of bioart and analyzes how ‘living art’ can be hampered by biosafety regulations. We argue that the exhibition and public discussion of bioart can lead to more informed societies.
|
|
Scooped by
mhryu@live.com
Today, 4:16 PM
|
Precise regulation of gene expression in batch bacterial cultures is challenging because the underlying dynamics vary with cellular physiological state over time. Although cell-silicon systems enable rapid, real-time optogenetic control, disturbance rejection remains difficult in batch culture because the plant dynamics shift across growth phases, limiting the effectiveness of fixed-gain controllers designed under constant-growth assumptions. Here, we present a multiscale model-guided feedback control framework for disturbance rejection in batch E. coli cultures. Frequency-response analysis shows that the input-output dynamics of gene expression depend strongly on growth phase, revealing operating-point-dependent limits on the disturbance rejection performance of a fixed-gain PID controller. To address this limitation, we develop two growth-aware control strategies: a gain-scheduled PID (PID-GS) controller that adapts to cellular physiological state, and a gain-scheduled feedback-feedforward controller (PID-GS-FF) that further compensates for growth perturbations. We also introduce a controller evaluation framework that identifies three distinct operating regimes for targeted experimental validation. Together, these results show that accounting for growth-state-dependent dynamics is necessary for robust disturbance rejection in batch culture and provide a control-oriented framework for regulating living systems with shifting operating conditions.
|
|
Scooped by
mhryu@live.com
April 5, 11:08 PM
|
Peptide hormones, which are biologically active short chains of amino acids (typically ranging from a few to about 100 residues), serve as crucial signalling molecules in plants. They play pivotal roles in regulating a wide spectrum of physiological and developmental processes through precisely regulated cell-to-cell communication networks. The discovery of plant peptide hormones has opened new frontiers in understanding growth regulation, developmental patterning, and defence signalling networks, offering significant biotechnological potential for the development of stress-resilient crops. Advances in genomics and receptor biology have uncovered the structural diversity and receptor-mediated perception of numerous peptide families, revealing their capacity to form dynamic regulatory circuits responsive to biotic and abiotic cues. In this review, we provide an updated synthesis of current knowledge on the structure, signalling mechanisms, and functional diversity of major plant peptide hormones, emphasising their emerging conceptual roles as information conduits that fine-tune systemic responses. Further discussion highlights how peptide receptor merges developmental plasticity with stress resilience through a feedback regulation, crosstalk with phytohormone signalling, and epigenetic control. Understanding these integrative peptides signalling networks provides new conceptual and translational avenues for emerging crop resilience and productivity under fluctuating environmental conditions.
|
|
Scooped by
mhryu@live.com
April 5, 12:06 PM
|
Metabolic engineering often treats microbial metabolism as an inventory of metabolites, reactions, and the enzymes that catalyze them. This Perspective argues that function emerges from metabolic architecture, the connectivities that bind reactions into stable regimes shaped, among other factors, by space and time. The Japanese Metabolism movement motivates an architectural view in which the same metabolites could lead to rather different phenotypes when cells reconfigure metabolic routing subjected to environmental constraints. Natural examples, including the native cyclic glycolytic wiring of Pseudomonas putida, show how redox supply and carbon flow depend on regime-level organization and space-influenced state changes. The same principles explain why microbial engineering often fails when intermediates leak, cofactors are misallocated, or timing breaks productive hand-offs. Serine-based synthetic cycles for one-carbon assimilation expose these limits as they must couple carbon entry, redox demand, and amino acid pool control around a chiral metabolite linked to translation. The emerging picture is that future designs should make routing, insulation, compartmentalization, and metabolic segregation explicit engineering targets.
|
|
Scooped by
mhryu@live.com
April 5, 11:22 AM
|
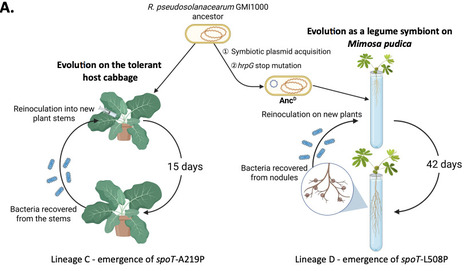
During evolution, bacteria have developed the ability to interact intimately with eukaryotic hosts. These interactions span a dynamic continuum ranging from pathogenicity to mutualism, along which bacteria can rapidly evolve and shift their lifestyles. However, the molecular mechanisms that enable bacteria to adapt to new hosts and to transition between distinct interaction modes remain poorly understood. Here, using a unique combination of two independent evolution experiments, we identified and characterized parallel adaptive mutations in spoT, which encodes the bifunctional (p)ppGpp synthetase-hydrolase. These mutations promote the adaptation of the plant pathogen Ralstonia pseudosolanacearum to two distinct plant-associated environments and two distinct lifestyles, the xylem of both susceptible and tolerant host plants as a pathogen and the root nodules of a legume as a symbiont, without compromising virulence on susceptible hosts. These mutations enhance the utilization of multiple carbon and nitrogen sources, including substrates known to be abundant in xylem sap, and increase bacterial exponential growth rate in minimal medium, suggesting reduced basal (p)ppGpp levels. Assessment of a strain deficient in SpoT synthetase activity confirmed that lowering basal (p)ppGpp levels is adaptive in both plant environments. Together, our findings reveal that fine-tuning intracellular (p)ppGpp concentrations represents an efficient strategy for optimizing bacterial adaptation to complex host-associated environments.
|
|
Scooped by
mhryu@live.com
April 5, 11:17 AM
|
Despite >50 years of methods development, specific antibodies are still generated at low throughput and remain in high demand across biotechnology. Most biologics and immunoprobes are monoclonal antibodies, developed using a combination of inoculating animals with a target antigen, engineered candidate libraries, and multiple rounds of selection using phage or yeast display. Here we introduce a synthetic biology scheme to eliminate the need for nearly all of these steps, by combining Surface display on E. coli and Phage display with the microvirus ΦX174, Assisting Continuous Evolution (SurPhACE). Instead of building libraries for screening, SurPhACE runs a closed evolutionary program. A typical experiment can have 1011 mutant candidates under active selection, with complete turnover of the mutant population every 30min, or >5x1012 unique mutants per day, using less than 100mL of bacterial culture media. We demonstrate SurPhACE for optimizing a nanobody to a related epitope, and develop novel nanobodies for an arbitrary target using a minimal starting library to establish a proof of concept and identify best practices for this scalable method for generating protein binders.
|
|
Scooped by
mhryu@live.com
April 4, 5:01 PM
|
Integrating semiconductors with microorganisms is attracting significant attention as a sustainable platform for solar-to-chemical conversion. This semibiological design combines the excellent light harvesting ability of semiconductor materials with intracellular biocatalytic pathways to enable efficient solar energy conversion into complex products with high selectivity. However, the effectiveness of this interdisciplinary biohybrid approach relies on a complex interfacial biotic–abiotic interaction, and it remains challenging to construct efficient and stable microbe–semiconductor systems for practical applications. In this review, we provide a systematic overview of the fundamental mechanisms behind microbe–semiconductor systems with an emphasis on interfacial electron transfer and highlight recent advancements in the assembly of biohybrids for solar-driven biosynthesis using nonphotosynthetic bacteria. First, we provide a comprehensive introduction of semibiological photosynthesis with an emphasis on extracellular electron transfer at the biotic–abiotic interfaces. Then, we discuss the engineering of biohybrid interfaces, the characterization of microbe–semiconductor interfacial electron transfer, and their deployment in solar-to-chemical conversion. We conclude by exploring the challenges in developing and optimizing biotic–abiotic interfaces as well as providing an outlook for potential future innovations. This review therefore presents the basic principles and provides guidance for the development of semibiological photosynthetic systems.
|
|
Scooped by
mhryu@live.com
April 4, 4:36 PM
|
Sliding-window phylogenetic analyses of multiple sequence alignments (MSAs) generate sequences of phylogenetic trees that can reveal recombination and other sources of phylogenetic conflict, yet comparing trees across genomic windows remains challenging. Phylo-Movies is a browser-based tool, also available as a standalone desktop application, that decomposes topological differences between consecutive phylogenetic trees into interpretable subtree migrations and animates these transformations. We demonstrate its utility in two contexts: identifying recombination breakpoints in norovirus genomes, where lineages shift from polymerase-based to capsid-based clustering at the ORF1/ORF2 junction, and detecting rogue taxa that change position across bootstrap replicates. Phylo-Movies complements summary statistics such as Robinson-Foulds distances by showing which lineages move, where they move from, and which new groupings they form. Phylo-Movies is freely available at https://github.com/enesberksakalli/phylo-movies, with a norovirus demonstration video at https://vimeo.com/1162400544, the first rogue taxon example at https://vimeo.com/1162561152, and the second example at https://vimeo.com/1162563101.
|
|
Scooped by
mhryu@live.com
April 4, 4:30 PM
|
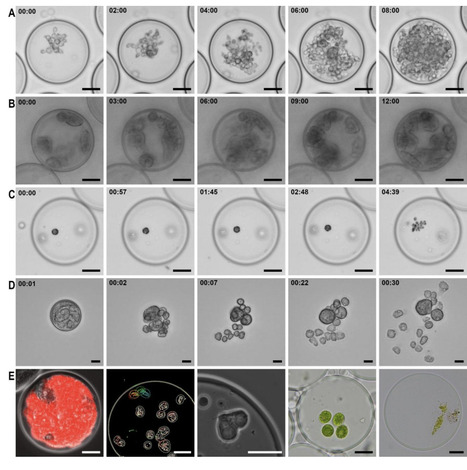
Semi-permeable capsules (SPCs) create enclosed porous microenvironments, diffusible to only small proteins and macromolecules. This presents a powerful tool for single cell observation, isolation, and sequencing. However, their range of use for sustaining viable microbial eukaryotes is largely unexplored. Single-cell eukaryotes are often understudied, with a wealth of unknown lifecycles, culturing methods and inter-microbial interactions, which are difficult to visualize. Here, we show that eukaryotes from eight different supergroups can be captured and propagated in SPCs. Encapsulation allowed observations of cell stages, motility and growth in a traceable and parallelized manner.
|
|
Scooped by
mhryu@live.com
April 4, 3:26 PM
|
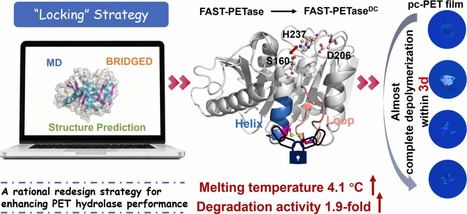
Extensive accumulation of polyethylene terephthalate (PET) plastic waste causes serious pollution to the global environment, and developing superior PET hydrolases is vital for the enzymatic degradation and biorecycling of PET. Here, we propose a “locking” strategy for the rational redesign of FAST-PETase, one of the best-performing PETase variants reported so far, to further improve its performance. The best variant, FAST-PETaseDC (A171C/S193C), exhibits 1.9-fold enhanced degradation efficiency compared with FAST-PETase at 50 °C, and a 4.1 °C increase in melting temperature (Tm). FAST-PETaseDC can almost completely depolymerize untreated post-consumer PET film within 3 d at 50 °C with periodic enzyme replenishment, two-fold faster than FAST-PETase. Molecular dynamics simulations reveal that the mutation locks Helix 5 and Loop 10 through a stable disulfide bond, and reduces the flexibility of the mutation sites and their connected regions. The structural changes consequently promote the substrate binding to the enzyme, facilitate the interaction within the catalytic triad, and rigidify the overall structure of the enzyme, leading to the improved degradation efficiency and thermostability. The study underscores the “locking” strategy as an effective way to enzyme redesign, and the engineered FAST-PETase variant is a promising hydrolase for the treatment and recycling of low-to-medium crystallinity PET plastic waste.
|























sweet, For sugars as signalling roles in plant–pathogen interaction, we refer readers to previous reviews (Bezrutczyk et al., 2018; Li et al., 2021; Morkunas and Ratajczak, 2014)