 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 7:56 PM
|
Prokaryotic Argonaute proteins (pAgos) are emerging as versatile tools in nucleic acid processing; however, their roles in bacterial physiology remain poorly defined. Here, we demonstrate that overexpression of Thermus thermophilus Argonaute (TtAgo) promotes transient bacterial filamentation in both T. thermophilus and E. coli through disruption of cell division checkpoints. Scanning electron microscopy and nucleoid DNA staining revealed defective septum formation and aberrant nucleoid segregation in filamentous cells. By observing the effect of truncated TtAgo variants on septum formation and nucleoid segregation in E. coli, we found that impairment of septum formation in the filamentous cells was independent of the DNA cleavage activity of the TtAgo protein. Further, we demonstrate that TtSSB interacts with TtAgo and recruits it to the replication fork, where it facilitates the DNA-binding activity of TtAgo. TtAgo acquires short DNA guides from broken double-stranded DNA and cleaves complementary chromosomal sequences, leading to DNA damage and filamentation. This filamentation triggers homologous recombination-mediated repair in T. thermophilus, allowing cells to return to a rod-shaped state. These findings reveal a mechanism by which the SSB-TtAgo interaction can modulate the bacterial cell cycle and DNA repair pathways, highlighting its potential for synthetic biology and biotechnology applications.
|
|
Scooped by
mhryu@live.com
Today, 6:41 PM
|
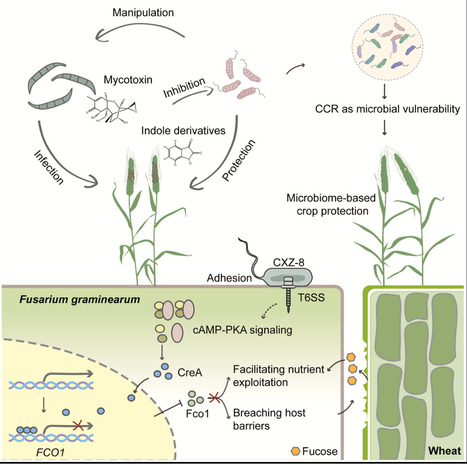
Carbon catabolite repression (CCR) acts as a switch, reprogramming nutrient utilization in fungal pathogens during the growth-to-colonization transition. However, whether this regulatory system can be exploited by other microbes remains unknown. Here, we demonstrate that Pseudomonas CXZ-8 attenuates the virulence of Fusarium graminearum by hijacking fungal CCR. CXZ-8 disrupts the infection-induced nuclear-to-cytoplasmic relocalization of the CCR master regulator FgCreA, thereby suppressing FCO1 expression, which is crucial for both host cell wall degradation and nutrient acquisition. This interference also benefits the bacterium by preventing the accumulation of host-derived indole derivatives and fungal mycotoxins that threaten its survival. Notably, approximately 20% of field-isolated bacteria exhibit similar FgCreA-stabilizing activity. Furthermore, we assembled a microbial consortium enriched for CCR-targeting bacteria, which conferred broad-spectrum disease resistance in field trials. These findings reveal a novel mode of interkingdom interference and establish CCR as a conserved microbial vulnerability, with implications for sustainable, microbiome-based crop protection.
|
|
Scooped by
mhryu@live.com
Today, 6:24 PM
|
The advent of ribosome profiling (an adaptation of RNA sequencing) to determine the translatome, has led to a huge improvement in our understanding of what parts of the transcriptome are translated. Many alternative open reading frames (ORFs) are now regularly being detected such as out-of-frame, overlapping, upstream or downstream reading frames, and alternative reading frames using non-canonical start codons. Various tools have been developed for the detection of such novel ORFs, but they lack the capacity to visually inspect reads—an important aspect of validation and prediction of translation. The integrated and visualization of ribosome profiling and RNA sequencing reads enables discrimination between transcriptional and translational signals, facilitating validation of predicted novel open reading frames. Furthermore, the inclusion of complementary evidence such as proteomic and long-read sequencing enables further validation of predicted novel open reading frames. Here, we present, InspectorORF (https://www.github.com/aylz83/inspectorORF), an R package that readily plots ribosome profiling reads, alongside RNA sequencing reads across transcripts and/or ORFs. Additionally, custom information can be plotted including data from additional conditions and samples, proteomic analyses and reads from long-read sequencing.
|
|
Scooped by
mhryu@live.com
Today, 6:16 PM
|
Understanding macromolecular structures of proteins and nucleic acids is critical for discerning their functions and biological roles. Advanced techniques—crystallography, nuclear magnetic resonance, and cryo–electron microscopy—have facilitated the determination of more than 180,000 protein structures, all cataloged in the Protein Data Bank. This comprehensive repository has been pivotal in developing deep learning algorithms for predicting protein structures directly from sequences. In contrast, RNA structure prediction has lagged and suffers from a scarcity of structural data. Here, we present the secondary structure models of 1098 primary microRNAs and 1456 human messenger RNA regions determined through chemical probing. We develop a deep learning architecture inspired from the Evoformer model of Alphafold and traditional architectures for secondary structure prediction. This model, eFold, was trained on our newly generated database and more than 300,000 secondary structures across multiple sources. We benchmark eFold on two challenging test sets of long and diverse RNA structures and show that our dataset and architecture contribute to increasing the prediction performance, compared to similar state-of-the-art methods. Together, our results reveal that merely expanding the database size is insufficient for generalization across families, whereas incorporating a greater diversity and complexity of RNA structures allows for enhanced model performance.
|
|
Scooped by
mhryu@live.com
Today, 6:02 PM
|
The treatment of gram-negative bacterial infections remains a formidable challenge due to their resilient outer membrane and adaptive evasion mechanisms. Herein, we present a multifunctional nanozyme hydrogel, copper telluride@cationic guar gum (CuxTe@CG), which acts as a gram-negative-specific bactericidal platform. This hydrogel integrates the unique enzymatic and physical properties of urchin-like CuxTe nanozymes with the biocompatible and adhesive cationic guar gum (CG) matrix. The CuxTe@CG hydrogel exhibits synergistic oxidase- and glutathione peroxidase-like activities, catalyzing the generation of reactive oxygen species (ROS) while depleting bacterial glutathione, thereby inducing lethal oxidative stress. Crucially, transcriptome sequencing revealed that the platform specifically targets Pseudomonas aeruginosa by down-regulating key genes involved in lipopolysaccharide (LPS) biosynthesis and flagellar assembly, compromising their primary defense and motility structures. This targeted interference with LPS and flagella amplifies the ROS-mediated attack, leading to enhanced and specific killing of gram-negative pathogens (E. coli, P. aeruginosa, and Klebsiella pneumoniae), effective biofilm disruption, and inhibition. In a P. aeruginosa-infected burn wound model, the CuxTe@CG hydrogel markedly accelerated healing by eliminating bacteria, promoting angiogenesis, and modulating inflammation, all while demonstrating excellent biosafety. This work establishes the CuxTe@CG hydrogel as a robust and targeted therapeutic strategy for combating stubborn gram-negative infections.
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
Transposons are a convenient vehicle to insert DNA into a bacterial genome, but widely-used transposons such as Tn7 are not able to hit custom targets. Recently, CRISPR RNA-guided integrases have been shown to direct the insertion of a mini transposon to a chosen site using a short guide sequence. We adapted this system for the widespread plant-associated genus Sphingomonas, revealing flexibilities and limitations of the tool. We uniquely tagged five genetically diverse strains, both in neutral sites and in sites with phenotypic consequences, and we demonstrate the utility of the tags for quantitative strain tracking in complex bacterial populations. Although we initially focused on the attTn7 site recognized by Tn7 as an insertion site with minimal fitness effects, we discovered via a genus-wide search that this site disrupts potentially important genes in some strains. Therefore we validated an improved site for benign integration in Sphingomonas and used a construct with guides targeting this site to transform a heterogeneous Sphingomonas population, bypassing prior strain isolation. Using a novel rapid and economical transposon mapping method, we were able to identify correctly-tagged primary transformant colonies with novel genetic content, thus demonstrating a short cut towards the establishment of diverse tagged synthetic communities for the experimental study of bacterial natural variation.
|
|
Scooped by
mhryu@live.com
February 24, 11:45 PM
|
Operons are gene clusters controlled by a single promoter that enable coordinated translation from a single messenger RNA. Here we describe an expansion of the CyanoGate MoClo toolkit to assemble synthetic operons. The versatile CyanOperon system includes two Level 0 acceptor vectors for building interchangeable promoter-ribosome bind site (RBS) combinations and 15 Level 1 acceptor vectors for the hierarchical assembly and expression of up to six genes within a single operon. The system also allows for operon assembly into a self-replicating vector or for chromosomal integration by homologous recombination. To showcase CyanOperon, we assembled the violacein biosynthesis pathway as an operon and demonstrated violacein production in Escherichia coli. We then constructed a 20-part RBS library to examine how spacer length between the Shine-Dalgarno sequence and start codon affects translation in E. coli and the model cyanobacterium Synechocystis sp. PCC 6803. Lastly, we compared the expression of up to three operonic fluorescent markers following chromosomal integration or from a self-replicating vector in E. coli and Synechocystis sp. PCC 6803. The CyanOperon system is publicly available and can be readily integrated with other MoClo systems to accelerate the development of standardized operon assemblies.
|
|
Scooped by
mhryu@live.com
February 24, 11:10 PM
|
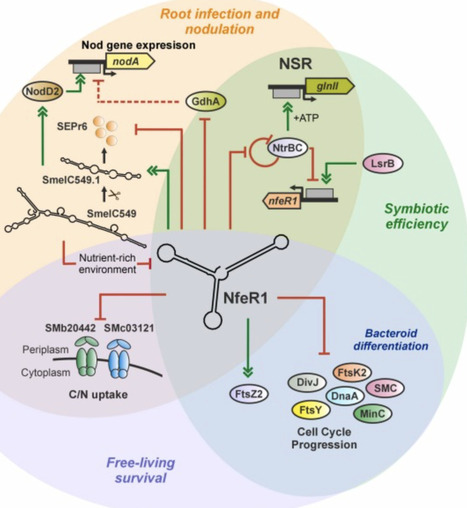
Bacterial small non-coding RNAs (sRNAs) remain understudied in the ecologically crucial nitrogen (N2)-fixing root-nodule Rhizobium-legume symbiosis. The only known rhizobial RNA regulator with broad symbiotic influence is the N-responsive trans-acting sRNA NfeR1, identified in the alfalfa symbiont Sinorhizobium meliloti. To pinpoint NfeR1 function, we profiled its RNA targets using MS2 affinity purification coupled with RNA sequencing (MAPS) in N stressed bacteria, a condition that drives nodulation. NfeR1 targets distinct regions of numerous mRNAs and sRNAs via three redundant anti-Shine-Dalgarno motifs, with downregulation constituting the primary regulatory outcome observed among the subset of validated targets. Target mRNAs span pathways differentially regulated throughout symbiosis, including N metabolism, motility, osmotolerance, and cell cycle control. Notably, NfeR1 modulates cell morphology and DNA replication by pervasive regulation of cell cycle mRNAs. It also silences gdhA, suggesting repression of glutamine dehydrogenase-dependent N assimilation, thereby promoting expression of nodulation genes, which is further fine-tuned by a novel RNA feedback loop involving NfeR1 and the dual-function sRNA SmelC549. Our findings position NfeR1 as a central hub within a structurally and functionally complex RNA network that coordinates N signaling and symbiotic performance in S. meliloti.
|
|
Scooped by
mhryu@live.com
February 24, 4:52 PM
|
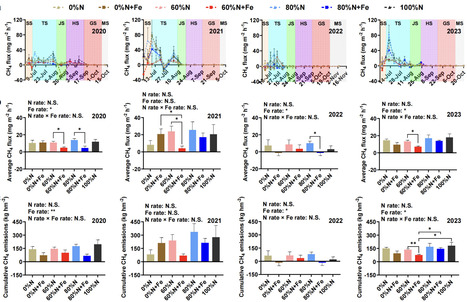
Methane (CH4) emissions from flooded paddy fields, exacerbated by excessive nitrogen (N) fertilizer application, trigger serious climate challenges. The impact of reducing N fertilization rate combined with iron (Fe) amendment on CH4 emissions remains unclear. This 4-year field study (2020–2023) investigated the effects of 100%, 80%, 60%, and 0% of the conventional N (urea and commercial organic manure) fertilization rate (100%N, 80%N, 60%N, and 0%N) as well as 80%, 60%, and 0% of the conventional N with the Fe powder (≥99% purity) amendment (80%N + Fe, 60%N + Fe, and 0%N + Fe) on CH4 emissions from subtropical rice paddies. The results revealed that 60%N + Fe treatments decreased cumulative CH4 emissions by 43.79% compared to the non-amended treatment, and by 57.33% in relative to the 100%N treatment in the 2023 rice season (P < 0.05). Meanwhile, Fe amendment significantly lowered the mcrA/pmoA ratio, which facilitated the decrease in CH4 emissions. Community assembly analysis showed that Fe amendment enhanced stochastic processes in methanogens at 60% of conventional N but reduced dispersal at 80% of conventional N, with opposite trends for methanotrophs. Co-occurrence networks demonstrated increased connectivity and reduced modularity under Fe amendment. Moreover, soil Fe2+ content and methanogen community structure, as critical drivers, were negatively correlated with CH4 flux and cumulative emissions (P < 0.05). Taken together, Fe amendment is a potent strategy to mitigate CH4 emissions under reduced N fertilization, offering a green production solution for global paddy systems.
|
|
Scooped by
mhryu@live.com
February 24, 4:37 PM
|
The small interfering RNA (siRNA) pathway directs broad-spectrum antiviral defense through RNA silencing so that virulent infection requires efficient suppression of the defense mechanism. Here, we show that strigolactone (SL) hormone signaling promotes antiviral silencing in rice plants by transcriptional activation of RNA-dependent RNA polymerase 1 (RDR1) and RDR6. We demonstrate that protein P3 of the rice grassy stunt virus (RGSV) blocks SL signaling by directly sequestering the receptor DWARF14 from DWARF3. Structural and functional analyses of the P3-DWARF14 complex reveal that the aspartic acid at position 102 (D102) of DWARF14 is essential for the P3 interaction but not for SL perception. Notably, a single D102N substitution of DWARF14, introduced into two rice cultivars by cytosine base editing (CBE) confers resistance against RGSV by blocking viral suppression of SL signaling-dependent antiviral silencing. Our findings establish a transgene-free strategy for engineering disease resistance by precise genome editing of the SL receptor to escape pathogen suppression of the endogenous defense pathway.
|
|
Scooped by
mhryu@live.com
February 24, 4:30 PM
|
Fungal plant pathogens have dynamic genomic architectures that can contribute to rapid evolution and adaptation to new niches. Zymoseptoria tritici, an important fungal pathogen of wheat, has a compartmentalized and rapidly evolving genome. In the genome of the reference isolate Z. tritici IPO323, 8 of the 21 chromosomes are accessory. In spite of the profound impact on genome organization, the origin of accessory chromosomes in Z. tritici is still poorly understood. By combining a multi-omics approach, we discovered an additional chromosome in Z. tritici isolates infecting wild grasses from the genus Aegilops, and we use this discovery to study the origin of accessory chromosomes. The newly identified chromosome presents characteristics similar to known accessory chromosomes in Zymoseptoria species, including presence-absence variation and enrichment with heterochromatin-associated histone methylation marks (H3K27me3). Interestingly, we found an orthologous chromosome in Zymoseptoria ardabiliae, a closely related fungal species also infecting wild grasses. This orthologous chromosome also presents accessory chromosome characteristics but lacks the enrichment of heterochromatin-associated methylation marks. Transcriptomic analyses revealed that the orthologous chromosome in Z. ardabiliae harbors active transposable elements (TEs) congruent with lower signatures of host-genome defense mechanisms against TE expansion and spread (quantified as repeat-induced-point mutation signatures). Our findings suggest that the chromosome has been exchanged between Z. tritici and Z. ardabiliae by introgressive hybridization events, underlining the relevance of hybridization in the evolution of new accessory chromosomes. We speculate that the regulation of TEs has not yet occurred on this new accessory chromosome in Z. ardabiliae, contributing to its rapid evolution.
|
|
Scooped by
mhryu@live.com
February 24, 4:17 PM
|
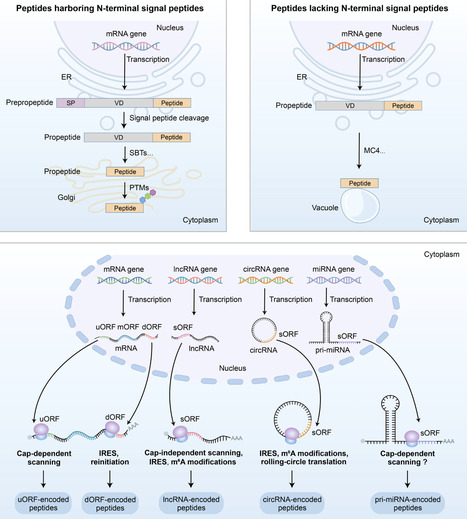
Plant peptides constitute a rapidly expanding class of signalling molecules essential to plant physiology, mediating key processes such as development, stress adaptation, and immune responses. This review traces the history of plant peptide research, from the seminal discovery of systemin to the recent identification of non-canonical peptides (NCPs) translated from small open reading frames (sORFs) in non-coding RNAs. We delineate the distinct biosynthetic pathways of canonical peptides (CPs), which undergo proteolytic processing and post-translational modifications, and NCPs, which are directly translated, often without further processing. The diverse biological functions of these peptides span development, reproduction, abiotic stress tolerance, biotic defence, and antimicrobial activity. Furthermore, we discuss emerging agricultural applications, including genetic engineering of peptides, exogenous peptide application, and trait optimization informed by natural peptide variation. Beyond agriculture, many plant peptides exhibit therapeutic potential due to their antimicrobial and anticancer properties. Despite significant advances, challenges remain in functional validation, field application, and scalable production. Future progress will depend on the integration of multi-omics approaches, artificial intelligence (AI)-driven prediction, and precision genome editing to fully harness the transformative potential of plant peptides for crop improvement and novel biopharmaceuticals.
|
|
Scooped by
mhryu@live.com
February 24, 12:57 PM
|
For a rapid and cost-effective evolution of tailor-made enzymes, we established a high-throughput in vitro selection platform named SMART (Single-Molecule Assay on Ribonucleic acid by Translated product), integrating mRNA display, next-generation sequencing, and bioinformatics. SMART represents a versatile system where a module termed an auxiliary unit allows enzyme-specific selection under various experimental conditions. Here, we report on the establishment of SMART for oxidases using a model enzyme, Schizosaccharomyces pombe d-amino acid oxidase (SpDAAO), and ascorbate peroxidase 2 as the auxiliary enzyme to detect hydrogen peroxide produced by the oxidase, and mediate biotinylation of active single-molecule display complexes. As a proof-of-concept, a library including site-saturation mutagenesis at the catalytic residue Y232 of SpDAAO was subjected to a single SMART selection round, yielding enrichment of the active enzyme variant. The results demonstrate the utility of SMART as a fast, robust, and efficient platform with the potential of customization for other enzyme chemistries through appropriate modifications of the auxiliary unit. Using SMART, desired enzyme variants can be selected in just a few hours by a single person without the need for costly equipment or any bias or limitations.
|
|
|
Scooped by
mhryu@live.com
Today, 7:50 PM
|
Fecal microbiota transplantation (FMT) has shown immunotherapeutic promise, yet its efficacy in non-small-cell lung cancer (NSCLC) remains unclear. We demonstrate that FMT improves anti-PD-1 efficacy and progression-free survival in a single-arm trial of advanced PD-L1-negative NSCLC. Analyzing over 2,000 metagenomes from diverse disease cohorts and healthy controls via a high-resolution strain-tracking framework, we reveal that phylogenetically distinct strains within identical species exert opposing therapeutic effects, resolving prior inconsistencies. We identify conserved ecological principles where engraftment relies on species-intrinsic metabolic and immune evasion traits. Crucially, successful colonization by specific beneficial strain variants correlates with positive clinical outcomes. Finally, we identify 38 priority species with robust engraftment potential and significant heterogeneity as candidates for precision therapeutics. These findings establish a strain-function-efficacy paradigm, elucidating the mechanistic basis of variable outcomes and guiding next-generation microbiome drug development.
|
|
Scooped by
mhryu@live.com
Today, 6:35 PM
|
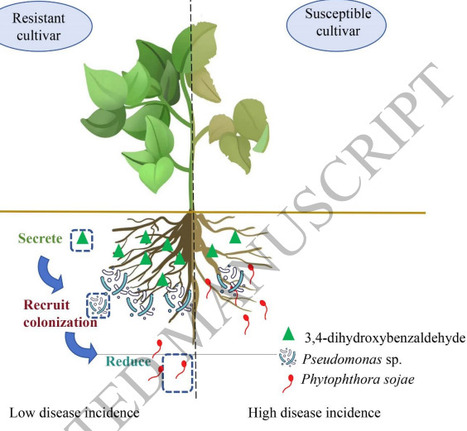
Rhizosphere microbiota mediate plant defense against soil-borne diseases, yet the mechanisms by which resistant soybean cultivars assemble protective microbiomes remain poorly understood. Using metagenomics, metabolomics, in vitro assays, and genetic approaches, we compared near-isogenic lines (Williams82, resistant; Williams, susceptible) to dissect plant–metabolite–microbe interactions mediating Phytophthora root rot (PRR) resistance. Transplanting rhizosphere soil from the resistant cultivar to susceptible plants significantly reduced PRR severity, correlating with Pseudomonas enrichment and accumulation of the key rhizosphere metabolite 3,4-dihydroxybenzaldehyde. We isolated a core beneficial strain, Pseudomonas parafulva ZY6, from the resistant rhizosphere. In vitro, 3,4-dihydroxybenzaldehyde treatment promoted ZY6’s biofilm formation, motility, and growth, while inhibiting Phytophthora sojae at higher concentrations. Knockout and overexpression of GmTL29 via hairy root transformation altered rhizosphere levels of 3,4-dihydroxybenzaldehyde, which in turn modulated the colonization of ZY6, the abundance of P. sojae, and the relative abundance of beneficial taxa such as Pseudomonas. Exogenous 3,4-dihydroxybenzaldehyde (0.1 μmol·g-1 soil) significantly reduced PRR disease index, increased rhizosphere bacterial diversity, and enriched Bacillus and Pseudomonas. Our study demonstrates that resistant soybeans shape a disease-suppressive rhizosphere, in which 3,4-dihydroxybenzaldehyde contributes as a prebiotic by selectively enriching beneficial microbes. These findings offer a metabolite-based strategy to engineer rhizosphere communities for sustainable soil-borne disease management.
|
|
Scooped by
mhryu@live.com
Today, 6:21 PM
|
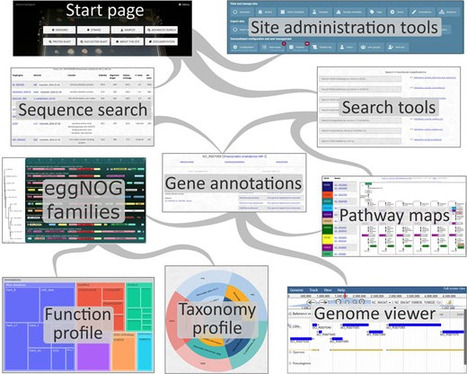
GenomeDepot is an open-source web-based platform for annotation, management, and comparative analysis of microbial genomic sequences and associated data including ortholog families, protein domains, operons, regulatory interactions, strain taxonomy, and sample metadata. GenomeDepot supports rapid creation of websites for user-defined genome collections that include bioinformatic tools for interactive genome browsing, Basic Local Alignment Search Tool (BLAST) search, annotation search, comparative genomic neighborhood visualization, and sequence download. Gene function annotations are generated by a customizable annotation pipeline. The pipeline runs annotation tools in Conda environments and can be easily extended with additional user-specified tools.
|
|
Scooped by
mhryu@live.com
Today, 6:10 PM
|
T7 RNA polymerase (T7 RNAP) is the most widely used enzyme for synthesizing therapeutic mRNA. However, RNA transcribed by T7 RNAP often contains double-stranded RNA (dsRNA) by-products that trigger innate immune responses and complicate purification. Here, we report an engineered T7 RNAP variant, M30, which exhibits higher catalytic efficiency and reduced dsRNA by-product formation. M30 was developed through 4 rounds of directed evolution using an ultrahigh-throughput aptamer-based fluorescence-activated droplet sorting system. M30 displays a 10-fold increase in catalytic efficiency over wild-type T7 RNAP at 37 °C, along with markedly enhanced thermostability and approximately 10-fold lower production of dsRNA by-products. mRNAs synthesized with M30 achieve efficient protein expression in human cells and in mice, while eliciting reduced immunogenicity compared with mRNAs produced by wild-type T7 RNAP. Biophysical assays and structural analyses suggest that these improvements result from increased DNA template binding affinity and decreased RNA binding affinity. Together, these features make M30 a promising catalyst for high-quality therapeutic mRNA production.
|
|
Scooped by
mhryu@live.com
Today, 12:13 AM
|
Conjugative plasmids drive bacterial evolution and antibiotic resistance spread, yet their gene expression must be silenced to protect the host. A histone-like protein H-NS represses many mobile and sedentary xenogenes but fails to silence the conjugal transfer vir operon of R6K, a prototype IncX plasmid. Instead, R6K encodes its own H-NS homolog, Sfx, to repress the vir operon. Here, we show that, unlike other plasmid silencers that target promoters, Sfx cooperates with Rho factor to arrest transcription elongation. ChIP-seq reveals that despite sharing similar DNA motifs and a preference for negative supercoiling, Sfx and H-NS occupy distinct niches: Sfx binds weakly to the chromosome but is enriched on the R6K vir operon, from which H-NS is excluded. We hypothesize that this selective targeting is mediated by Sfx-vir interactions and phase separation. We show that Sfx binding to vir DNA critically depends on DNA topology but not on the target location. Our results suggest that Sfx phase separates with R6K to ensure its preferential recruitment to the plasmid DNA and forms stable nucleoprotein filaments that are impermeable to competitors. These findings reveal how histone-like proteins can partition the genome into distinct regulatory niches, a strategy likely mirrored across all life.
|
|
Scooped by
mhryu@live.com
February 24, 11:47 PM
|
Biofilm communities exhibit emergent properties that exceed the sum of contributions from individual members of the community. Here, we describe a multilayered metabolic interaction that drives enhanced biofilm formation among three bacterial species from the plant rhizosphere. Comparative metatranscriptomic and metabolomic analyses reveal that Bacillus velezensis-secreted 5-aminovaleric acid promotes the growth of the other community members, Burkholderia contaminans and Acinetobacter baumannii. In return, B. contaminans supplies branched-chain amino acids for B. velezensis. Branched-chain amino acids and cell-cell signaling acyl-homoserine lactones from B. contaminans induce biosynthesis of the siderophore bacillibactin in B. velezensis, that is further enhanced by A. baumannii. In exchange, the B. velezensis-secreted siderophore promotes the growth of B. contaminans in iron-limited conditions, which benefits the multispecies biofilm community in vitro and promotes plant growth performance in iron-depleted soil. Our study reveals the molecular mechanisms underlying an emergent rhizosphere biofilm community function and demonstrates its importance in plant-microbe interactions.
|
|
Scooped by
mhryu@live.com
February 24, 11:19 PM
|
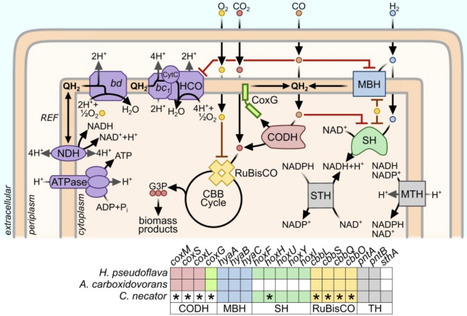
Some aerobic bacteria can convert syngas, an energy-dense mixture of CO, H2, and CO2 derived from waste gasification, into platform chemicals and products such as single-cell protein and bioplastics. Despite the enormous theoretical energy yield of aerobic syngas conversion, few cultured bacteria can mediate this process, and none do so quickly or efficiently. This reflects the dual challenges that known pathways of aerobic CO conversion are highly inefficient, and most H2-oxidizing enzymes are highly CO-sensitive. Here, we propose three strategies to overcome these challenges: evolving and engineering existing syngas-converting strains, isolating novel syngas-converting microbes from gas-rich environments, and introducing CO-insensitive hydrogenases and direct CO conversion pathways into industrial chassis strains. If this can be achieved, efficient aerobic syngas conversion would become a cornerstone of a sustainable bioeconomy.
|
|
Scooped by
mhryu@live.com
February 24, 4:56 PM
|
A field-deployable DNA sequencing approach for quantitative microbial community profiling can enable rapid responses for a range of applications in the water sector—from process control to wastewater surveillance. Current quantitative approaches require complex instrumentation and have long turnaround times for DNA recovery and absolute quantitation. In this study, we report a field-deployable rapid detection and rapid absolute quantitation (rD+rQ) workflow that leverages real-time Nanopore sequencing for quantitative metagenomics. This workflow integrates a high-molecular-weight DNA recovery protocol for diverse environmental matrices of relevance to the water sector, and multiplexed Nanopore sequencing with barcoded spike-in-based calibration (BSINC). BSINC using multispecies genomic spike-in controls exhibits significantly higher calibration accuracy compared to conventional approaches that utilize either a single DNA fragment or single organism spike-in controls. Dynamic detection and quantitation limits were established based on the coverage fraction of sequenced genomes and the coefficient of variation of genome copy numbers across replicates to enhance the accuracy and precision of microbial quantitation. The rD+rQ workflow achieves species-level identification and absolute quantitative results comparable to digital PCR in environmental samples. This portable laboratory and easy-to-use rD+rQ workflow should facilitate rapid decision-making for the water industry.
|
|
Scooped by
mhryu@live.com
February 24, 4:48 PM
|
Microbiology research often requires tracking of specific bacterial strains within a host infection system or in the environment, as well as differentiation of strains in a co-infection or microbe-microbe interaction scenario. Various tools are used for this purpose, including antibiotic resistance marker genes, fluorescent proteins, DNA sequence-based methods, and phenotypic markers. Chromoproteins produce intense pigmentation visible in ambient light and are a unique option for bacterial tracking that does not require use of antibiotics, specialized equipment, or DNA sequencing. Development of traceable bacterial strains across a wide range of species is important to facilitate the investigation of challenging research questions and expand our understanding of microbial dynamics in complex environments. In this study, different species of plant pathogenic bacteria (Xylella fastidiosa, Pantoea stewartii, Pseudomonas syringae, and Xanthomonas campestris) were modified with a set of chromoproteins and tested in plant infection assays to evaluate chromoprotein stability and impact on bacterial pathogenicity. The primary goal of this study was to develop chromoprotein-modified strains of X. fastidiosa using chromosomal insertion, which was highly successful and stable during infection in grapevines. Plasmid-based expression of chromoproteins in P. stewartii, P. syringae, and X. campestris had mixed results depending on the specific species-chromoprotein combination. Overall, these results provide some successful chromoprotein-modified plant pathogen strains for use by the research community, as well as insight into which chromoproteins might be best utilized in different bacterial species.
|
|
Scooped by
mhryu@live.com
February 24, 4:33 PM
|
Microbial-induced calcium carbonate precipitation (MICP) has emerged as a promising biotechnological approach for addressing coal dust pollution in mining and industrial environments. Among the various biological agents, urease-producing bacteria play a central role in catalyzing urea hydrolysis, leading to the generation of carbonate ions that react with calcium to form calcium carbonate (CaCO3). This biologically formed mineral binds dust particles, enhances surface stability, and reduces airborne pollutant dispersion. While MICP presents clear environmental and structural advantages, including low toxicity, long-term ecological compatibility, and compatibility with natural ecosystems, the underlying mechanisms, particularly the microbial adhesion to coal particles and subsequent mineralization dynamics, remain poorly understood. High production costs, sensitivity to environmental conditions, and lack of large-scale validation have also limited the practical implementation of microbial dust suppressants. This review provides a comprehensive look at the current research on the biological processes and application strategies of MICP in coal dust suppression, emphasizing the role of ureolytic bacteria, carrier systems, and calcium sources. Furthermore, it explores recent advancements in microbial strain selection, additive incorporation, and delivery methods that aim to optimize microbial survival and mineralization efficiency in real-world mining conditions. Future perspectives are discussed to support the development of cost-effective and scalable microbial formulations, paving the way for green and durable solutions in mine dust management.
|
|
Scooped by
mhryu@live.com
February 24, 4:23 PM
|
Mycorrhizal fungi represent one of the oldest and most successful symbioses in plant evolution. Communication among mycorrhizal fungi and plants occurs prior to direct contact among them through different and variable biochemical signals, including microRNAs, hormones, small peptides and volatile organic and inorganic compounds. Volatile organic compounds (VOCs) emerge as key chemical signals that enable the transmission of chemical messages modulating plant and microorganism responses in both below- and above-ground ecosystems. The diversity and concentration of mycorrhizal VOCs will vary depending on the environment and the emitting organism and are usually related to changes in the conformation of root architecture and lateral root formation mediated by auxin and strigolactones. Moreover, the study of the effects of mycorrhizal VOCs in the tolerance to abiotic and biotic stress are still scarce although there are some promising results pointing out to the effect of these VOCs in plant development under osmotic stress conditions, and their properties as antifungal and antibacterial molecules. However, the information regarding the molecular mechanisms involved in mycorrhizal VOCs signaling and their effect on plants remains still elusive. The understanding of VOC-mediated plant-mycorrhizal interactions, together with the technical improvements for their detection and mode of application in the field, will open new avenues for biotechnological crop improvement and management that not only will reduce the dependence on agrochemicals but also fosters soil health and plant resilience.
|
|
Scooped by
mhryu@live.com
February 24, 4:04 PM
|
The plant microbiome plays a central role in regulating plant health and resilience, providing eco-friendly alternatives to agrochemicals. Plant-associated Streptomyces species are prolific producers of structurally diverse natural products with a demonstrated role in promoting plant growth. Coumarins are prevalent plant metabolites that shape the root microbiome, but their impact on microbial natural product biosynthesis is poorly understood. Here, we demonstrate that the coumarins scopoletin and its glucoside scopolin remodel specialized metabolism in the Arabidopsis root endophyte Streptomyces sp. ATMOS53. Multiomics analyses revealed that the coumarins activate the biosynthesis of the pyrrolizidine alkaloids bohemamines and alter the balance in anthracycline biosynthesis, with reduced production of late-stage anthracycline congeners and accumulation of shunt metabolites earlier in the pathway. These metabolic shifts resulted in a marked reduction of the antimicrobial activity of ATMOS53 against plant-associated Bacillus and Paenibacillus species. Notably, coumarin-mediated repression of anthracycline production was also observed in the established producers Streptomyces peucetius and Streptomyces galilaeus, indicating that the regulatory effect on anthracycline biosynthesis is conserved in streptomycetes. Our findings highlight coumarins as modulators of specialized metabolism of Streptomyces and show the significance of plant-derived chemicals for the control of the biosynthetic capacity of plant-associated microbes.
|




























droplet, 3st idea