 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:24 PM
|
Three different cultures of cyanobacteria: Synechocystis sp., Leptolyngbya sp., and a co-culture of both Synechocystis sp. and Synechococcus sp. were tested for their polyhydroxybutyrate (PHB) production potential under diverse conditions. High-throughput screening in a multi-well plate, using Nile blue fluorescence to estimate PHB accumulation, enabled simultaneous testing of multiple parameters without complex setups and faster PHB estimation compared to conventional gas chromatography. Fluorescence results indicated that the best conditions for PHB accumulation were nitrogen depletion under darkness at 30°C with acetate supplementation after 4 days of glycogen accumulation. Finally, these optimal conditions were validated in single-batch proof-of-concept photobioreactors (1 L working volume). The scale-up proved successful, yielding 7% PHB dry cell weight (dcw) for Synechocystis sp. and a promising 13% PHB dcw for Leptolyngbya sp. In addition, scale-up experiments in 1-L photobioreactors demonstrated that a dedicated glycogen pre-accumulation step is unnecessary, as PHB can be efficiently synthesized directly from acetate.
|
|
Scooped by
mhryu@live.com
Today, 5:10 PM
|
Indigoidine is a blue pigment biosynthesized by a single-module Non-Ribosomal Peptide Synthetase (NRPS) using L-glutamine as substrate. Despite its potential as a colorimetric reporter, no such system has been established from it to date. We used a recently characterized interdomain fusion site located between its adenylation (A) and thiolation (T) domains to develop the Indi2GO system, which provides a naked-eye detectable and quantitative optical readout of transient and covalent protein-protein-interaction (PPI) in living cells. Indi2GO enables high-throughput benchmarking and optimization of PPI tools in a standard 96-well plate reader format, without requiring exogenous substrates, specialized equipment or complex analytical workflows. We demonstrate its broad applicability with three widely used protein-protein interaction tools: SYNZIPS, inteins, and the SpyTag:SpyCatcher system. We used Indi2GO to validate novel SYNZIP pairs, which we used in NRPS engineering, highlighting its applicability for the development of novel PPI-mediating tools in the context of NRPS engineering and synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 3:19 PM
|
Nanoparticles are now central to many drug and vaccine delivery strategies, but most require reformulation for each application. Bacteriophage‑derived nanoparticles offer a genetically encoded, structurally defined, and modular alternative. This review organizes recent advances along three tunable axes — scaffold, surface, and cargo — and highlights hybrid phage–polymer/lipid/inorganic constructs that expand stability, targeting, and loading. We survey applications from multivalent vaccines and oversized gene transfer to precision microbiome editing, and outline translational hurdles. Phage-derived products are approaching translation, with virus-like particle-based vaccines and CRISPR-enhanced antimicrobial phages in clinical trials. Finally, we preview emerging opportunities, including AI‑guided capsid and receptor‑binding protein design, cell‑free phage synthesis, and standardized ‘reference’ phage chassis that can be combined with traditional nanoparticles, positioning phage nanoparticles as reusable, plug-and-play nanomedicines.
|
|
Scooped by
mhryu@live.com
Today, 2:38 PM
|
In contrast to the rod shape at 37°C, the morphology of E. coli cells at temperatures just above the minimum temperature of growth is small rods. A study was initiated to determine the requirement of nucleoid-associated protein FIS for growth and genome compaction in the small rods at low temperature. Growth and nucleoid staining analyses revealed that the fis null mutant displayed decreased growth and initially formed filaments containing decondensed nucleoids at 12°C, indicating that FIS facilitates production of small rods with condensed nucleoids at low temperature. However, characterized by biphasic growth at low temperature, the fis null mutant exhibited increased growth, cell division, and nucleoid condensation following an acclimation phase. Therefore, the absence of FIS with nucleoid decondensation leads to an adaptation mechanism, termed FIS Null Adaptation Response, that causes a shift towards nucleoid condensation resulting in genome compaction in small rods. Furthermore, overproduction of the HsIVU protease suppressed the cold-sensitive phenotypes of the fis null mutant indicating that degradation of a natural substrate of the protease alleviates the requirement of FIS at low temperature. In addition, null mutations of genes encoding natural substrates of HsIVU (exoribonuclease RNAse R, and cell division inhibitor SulA) were identified as extragenic suppressors of the fis null mutation.
|
|
Scooped by
mhryu@live.com
Today, 2:26 PM
|
Simultaneously introducing diverse genomic edits remains a challenge in crop genome engineering. Here we describe a twin prime editing-based knockout (TKO) system that installs stop codon clusters (SCCs) for precise translational termination with minimal in-frame mutations. TKO achieves knockout efficiencies of up to 70.5%, 58.6% and 75.1% in rice, maize and wheat protoplasts, respectively, and produces heritable knockout alleles in 96.8% of regenerated rice plants. In hexaploid wheat, TKO outperforms Cas9 4.2-fold in generating triple-homolog knockouts, largely by reducing in-frame mutations. Orthogonal TKO editors with sequence-divergent SCCs enable simultaneous knockout of up to ten genes without cross-interference. Integration of TKO with conventional prime editing establishes TRIM1 (TKO editor-enabled gene rupture and development of integrated multitype genome modification system) for simultaneous knockout and precise editing, achieving a 22.8% coediting of four genes in rice. TRIM2 extends this capacity to kilobase-scale modifications through a prime editor–recombinase system, enabling a 4.9-kb insertion (1.2% efficiency) and gene knockout (up to 79.8%) in protoplasts. Plant genome editing is multiplexed with twin prime editing.
|
|
Scooped by
mhryu@live.com
Today, 12:17 AM
|
Understanding protein functions is crucial for interpreting microbial life; however, reliable function annotation remains a major challenge in computational biology. Despite significant advances in bioinformatics methods, ~30% of all bacterial and ~65% of all bacteriophage (phage) protein sequences cannot be confidently annotated. In this review, we examine state-of-the-art bioinformatics tools and methodologies for annotating bacterial and phage proteins, particularly those of unknown or poorly characterized function. We describe the process of identifying protein-coding regions and the systems to classify protein functionalities. Additionally, we explore a range of protein annotation methods, from traditional homology-based methods to cutting-edge machine learning models. In doing so, we provide a toolbox for confidently annotating previously unknown bacterial and phage proteins, advancing the discovery of novel functions and our understanding of microbial systems.
|
|
Scooped by
mhryu@live.com
June 4, 11:58 PM
|
Harnessing the innate growth, self-repair, and adaptive capabilities of living systems within engineered devices could transform static buildings via domestic infrastructures into dynamic, self-sustaining platforms. Electroactive biofilms (EABs) provide a unique interface for this vision, naturally converting organic matter into electricity, treating wastewater, and processing complex information. Recent breakthroughs in synthetic biology and artificial intelligence now allow EABs to be programmed as biologically intelligent components—such as living transistors and logic processors—rather than simple biocatalysts. This opinion article outlines a roadmap for transitioning EAB-enabled hybrid biological-artificial systems from laboratory prototypes into integrated architectures for decentralised resource recovery. Ultimately, these bio-intelligent technologies enable a circular economy in which buildings function as metabolic organisms, redefining our relationship with the built environment.
|
|
Scooped by
mhryu@live.com
June 4, 11:38 PM
|
Biosynthetic needs of a cell and the generation of precursors and energy in catabolic reactions must be faithfully balanced. Even in model organisms such as Bacillus subtilis, there are still important gaps in our knowledge. The key bottleneck in research is the lack of novel research hypotheses. New concepts and methodologies can help to develop such hypotheses. Here, we discuss how the introduction of proteome-wide protein-protein interaction mapping by in vivo cross-linking, the AI-mediated prediction of structure models for each protein, and the possibility to compare those models highly efficiently aid the development of novel hypotheses. Moreover, the focused use of suppressor screens can help to get new unbiased insights. We demonstrate how these approaches are applied to B. subtilis. Global cross-linking combined with the power of AI provided a testable hypothesis to unravel the long-standing open question of how iron is sensed in B. subtilis and related bacteria. This is of particular importance as iron is the growth-limiting factor for most bacterial pathogens. The isolation of suppressor mutants that are resistant to growth-inhibiting amino acids has identified novel amino acid exporters. Importantly, the corresponding genes belong to the most poorly expressed genes in B. subtilis, and they are only activated under selective pressure by mutations that affect corresponding transcription factors or the promoter regions of the exporter genes. As the approaches discussed here have only recently been brought to wide application, we can expect that they will be very fruitful in gaining a better understanding of metabolism and metabolic homeostasis.
|
|
Scooped by
mhryu@live.com
June 4, 11:20 PM
|
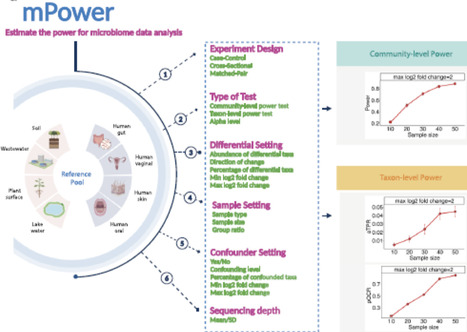
Power analysis is a critical step in designing a microbiome study. Existing power calculation tools for microbiome studies mainly rely on parametric models of the sequencing counts, which underestimate the complexity of microbiome data and could produce overly optimistic power estimates. In this work, we present a new simulation-based power analysis tool, mPower, for microbiome study design. The tool uses a real data-based semi-parametric simulation framework to generate realistic microbiome data, upon which the power assessment is performed. Coupled with a select differential analysis tool, our power tool supports different study designs, including cross-sectional, case-control, and matched-pair studies, with or without confounders. It allows power analysis for both community-level and taxon-level testing. By using microbiome reference datasets from different environments, the users could perform power calculation based on the environment of interest. The mPower is primarily designed for 16S amplicon sequencing data, and it also incorporates a parametric simulation framework that enables power analysis for shotgun metagenomic data. We showcase the application of mPower with several real-world examples. The web interface of mPower is available at https://microbiomestat.shinyapps.io/mPower/. Video Abstract
|
|
Scooped by
mhryu@live.com
June 4, 11:07 PM
|
Natural products (NPs) remain vital to drug discovery, yet the identification of novel bioactive NPs is frequently hampered by the rediscovery of known compounds in traditional screening and the “bioactivity gap” in genome mining. Self-resistance-guided genome mining has emerged as a transformative strategy to address these challenges, leveraging co-localized resistance genes within biosynthetic gene clusters (BGCs) to predict NPs’ molecular targets. This review summarizes recent progress in discovering novel NPs that target essential cellular processes, including protein synthesis, protein degradation, DNA integrity, and primary metabolism. We further highlight key technologies and strategies designed to accelerate this discovery workflow and discuss the limitations and opportunities of self-resistance-guided genome mining for the systematic discovery of precision therapeutics in the genomic era.
|
|
Scooped by
mhryu@live.com
June 4, 10:52 PM
|
Phage therapy often fails when bacteria evolve resistance. We argue that phage selection should begin with receptors whose modification imposes predictable costs, turning resistance into reduced virulence, antibiotic resensitization, or other exploitable trade-offs. Receptor-constrained evolutionary traps offer a framework for designing phages that steer—not merely suppress—bacterial evolution effectively.
|
|
Scooped by
mhryu@live.com
June 4, 5:02 PM
|
In recent years, synthetic biology has been widely applied to engineer and program cellular behaviors. Using this approach, bacteria can be designed to express immunotherapeutic agents, improve tumor targeting, and deliver therapeutic payloads directly to tumor sites. To further improve efficacy, strategies such as hypoxia-responsive promoters, bacterial swarming, and extracellular vesicles (EVs) have been investigated, along with the synergistic effects of combining bacterial therapy with other treatments (e.g., photodynamic therapy, chemotherapy, immune checkpoint inhibitors). This review summarizes recent advances in synthetic biology for bacteria-based cancer immunotherapies, focusing on how bacterial agents activate the immune system and the engineering strategies used to achieve tumor targeting.
|
|
Scooped by
mhryu@live.com
June 4, 4:57 PM
|
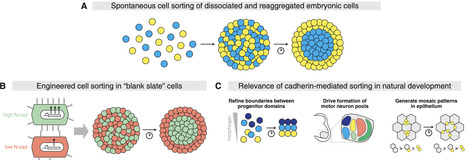
Developmental biology seeks to understand how multicellular organization emerges from cell-cell interactions. Advances in stem cell and synthetic biology now enable researchers to rebuild developmental processes outside the embryo, with varying degrees of resemblance to natural systems. While some reconstituted systems reveal how development occurs, others uncover what is possible. This perspective examines how such bottom-up approaches have elucidated general principles and causal mechanisms of multicellular organization. We argue that synthetic systems, though simplified, provide powerful platforms to test the limits of developmental potential, disentangle causal relationships, and inform predictive models. With rapid advances in genomic engineering, imaging, and computational modeling, leveraging these engineered systems to discover what is possible holds transformative promise for understanding what is happening in nature.
|
|
|
Scooped by
mhryu@live.com
Today, 5:16 PM
|
Predicting protein-ligand complex structures is a central challenge in drug discovery. While recent co-folding models such as AlphaFold-3 achieve accurate structure prediction, they fail to generalize to underexplored binding interfaces - systematically misplacing ligands, particularly for allosteric or structurally novel targets. To address this gap, we present ACER (A daptive Co-folding via pocket E xploration and pose R anking), a training-free framework that (a) enables co-folding models to systematically explore alternative binding pockets, and (b) leverages the discovered pockets to increase pose accuracy. Our method enables the efficient discovery of non-prevalent pockets without prior expert knowledge. ACER improves pocket discovery and pose accuracy on allosteric targets and structurally novel complexes, successfully modeling binding interfaces that are under-represented or absent from the training set. Our results demonstrate how improved sampling dynamics enhance the generalizability of co-folding models without retraining.
|
|
Scooped by
mhryu@live.com
Today, 4:56 PM
|
The widespread emergence of antibiotic-resistant pathogens poses a significant global health challenge and underscores the need for novel approaches to accelerate antimicrobial discovery. Antimicrobial peptides (AMPs) have gained attention as promising candidates due to their broad-spectrum activity, including efficacy against multidrug-resistant bacterial strains. SAJO-2, an antimicrobial peptide developed by Sarojini and colleagues, features a tryptophan zipper-like motif incorporating a central d-Phe-2-Abz unit, where 2-Abz functions as a conformationally constrained β-turn-inducing peptidomimetic scaffold. Modification of SAJO-2 in prior joint work from our groups through differential fluorination enhanced its antimicrobial potency; however, it also increased susceptibility to enzymatic digestion by β-trypsin. To address this limitation, the current research focuses on improving the overall efficacy of SAJO-2 through the incorporation of D-amino acids, beta backbone modifications, and a bulky pentafluorinated amino acid residue. All modified peptides exhibit resistance to enzymatic degradation, while antimicrobial activity was retained to differing degrees across organisms.
|
|
Scooped by
mhryu@live.com
Today, 3:03 PM
|
Nanoparticles are now central to many drug and vaccine delivery strategies, but most require reformulation for each application. Bacteriophage‑derived nanoparticles offer a genetically encoded, structurally defined, and modular alternative. This review organizes recent advances along three tunable axes — scaffold, surface, and cargo — and highlights hybrid phage–polymer/lipid/inorganic constructs that expand stability, targeting, and loading. We survey applications from multivalent vaccines and oversized gene transfer to precision microbiome editing, and outline translational hurdles. Phage-derived products are approaching translation, with virus-like particle-based vaccines and CRISPR-enhanced antimicrobial phages in clinical trials. Finally, we preview emerging opportunities, including AI‑guided capsid and receptor‑binding protein design, cell‑free phage synthesis, and standardized ‘reference’ phage chassis that can be combined with traditional nanoparticles, positioning phage nanoparticles as reusable, plug-and-play nanomedicines.
|
|
Scooped by
mhryu@live.com
Today, 2:31 PM
|
Cardioprotective effects of current therapies for mitigating ischaemia/reperfusion (I/R) injury have had limited success. The major challenge is to effectively control oxidative stress while preserving mitochondrial function in a timely manner. Hydrogen (H2) selectively reduces cytotoxic oxygen radicals, aiding in the regulation of physiological and pathological functions. However, the efficacy of H2 therapy is highly dependent on the amount and rate of H2 release, making it critically important to develop rapid, simple and efficient techniques for evolving therapeutic H2. Here we encapsulate H2-producing photosynthetic bacteria (PSB) in an injectable porcine dermal extracellular matrix (ECM) hydrogel to facilitate cardiac I/R injury repair. Upon light exposure, sustained and high H2 production from PSB hydrogel preserves mitochondrial homeostasis and essential functions. In a porcine model of cardiac I/R injury, PSB hydrogel treatment effectively mitigates myocardial damage and salvages jeopardized myocardium. We anticipate that this bacterial therapy for photosynthetic H2 production could provide an improved treatment for I/R-related diseases. Photosynthetic bacteria embedded in an injectable hydrogel produce ROS-scavenging species for mitigating myocardial damage and improving heart repair in a porcine model.
|
|
Scooped by
mhryu@live.com
Today, 12:37 AM
|
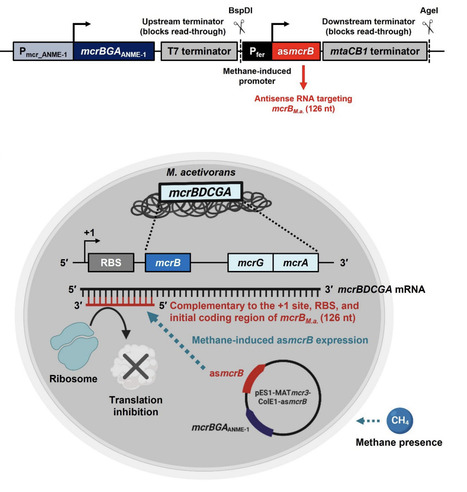
Utilizing methane and carbon dioxide before it can enter the upper atmosphere is beneficial for mitigating climate change as well as for producing valuable chemicals. Because anaerobic methanotrophic archaea (ANME) have not yet been cultured in isolation, we previously reversed methanogenesis by cloning the genes encoding methyl-coenzyme M reductase (Mcr) derived from Black Sea ANME-1 into the methanogen Methanosarcina acetivorans. The resulting engineered archaeal strain captures, rather than produces, methane and may be used to convert methane and carbon dioxide into electricity, acetate, L-lactate, and ethanol. However, the engineered M. acetivorans strain also contains a chromosomal locus encoding its native Mcr (McrM.a.), which produces methane from substrates such as methanol, whereas the heterologously expressed ANME-1 Mcr (McrANME-1) promotes methane oxidation. Therefore, we reasoned that McrM.a. may compete with McrANME-1-mediated reversal of methanogenesis. To enhance the reversal of methanogenesis, here we implemented an antisense RNA (asRNA) silencing approach to suppress McrM.a. during growth on methane while still allowing its expression during routine growth on methanol. We found that silencing McrM.a. during McrANME-1-mediated growth on methane increased ethanol and acetate production by more than an order of magnitude. These results were corroborated by both a more than 10-fold increase in methane utilization by McrANME-1 and a greater than 1,000-fold reduction in the McrM.a. mcrBGA transcript levels under methane-grown conditions. Therefore, asRNA-mediated silencing may be used to enhance methane capture by suppressing production of the host McrM.a. for biotechnological applications.
|
|
Scooped by
mhryu@live.com
Today, 12:10 AM
|
Mushroom-forming Agaricomycete fungi underpin global nutrient cycling and carbon sequestration, and support large and growing markets across food, medicinal supplements, and biomaterials. Yet most commercial and research uses still rely on wild-type strains, highlighting the opportunity for genetic engineering to expand possibilities for both fundamental research and biotechnological applications. In this review, we highlight progress toward synthetic biology in Agaricomycetes, and outline the main barriers that limit predictable genetic engineering. We emphasize engineering constraints unique to mushroom biology, including complex sexual cycles, heterokaryosis, and strain instability during transformation and outgrowth. We then transition to gene expression bottlenecks: the scarcity of characterized promoters and terminators, the challenges for gene integration posed by the condensed nature of Agaricomycete genomes, and the effects of introns and specific sequence motifs. Finally, drawing inspiration from progress in related fungi and other eukaryotes, we highlight the priorities for the field: systematic cross-species evaluation of genetic parts, development of more sophisticated gene-editing strategies, higher-throughput screening methods, and the establishment of a unifying model system. These advances would enable new possibilities in the study and use of Agaricomycetes, establishing these elusive organisms as programmable platforms for sustainable biomanufacturing, designer biomaterials, climate solutions, and mechanistic studies of fungal biology.
|
|
Scooped by
mhryu@live.com
June 4, 11:48 PM
|
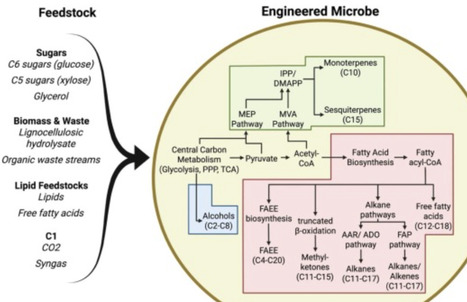
Sustainable aviation fuels (SAF) are critical for decarbonising the hard-to-abate aviation sector, which significantly contributes to global CO2 emissions. Conventional SAF production routes, such as Hydroprocessed Esters and Fatty Acids, Fischer-Tropsch and Alcohol-to-Jet, offer drop-in compatibility but are constrained by feedstock availability, high costs and environmental impacts. This review highlights, as promising alternatives, microbial bioproduction via precision fermentation of SAF-relevant compounds from low-cost feedstocks, with reduced land use and enhanced circularity. Here, we focus on microbially derived SAF precursors such as alcohols, terpenes, fatty acid ethyl esters, methyl ketones and saturated hydrocarbons, as well as recent advances in host engineering, pathway design, and bioprocess optimization.
|
|
Scooped by
mhryu@live.com
June 4, 11:27 PM
|
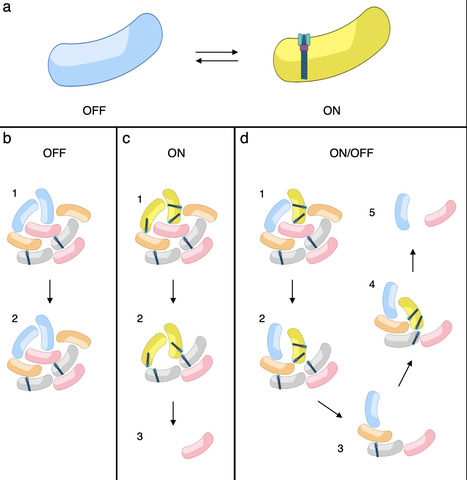
The type VI secretion system (T6SS) is a widespread nanoweapon deployed by bacteria to eliminate competitors in polymicrobial environments, allowing niche colonization or host invasion. Fluorescent microscopy recordings have shown that T6SS expression and/or activation is heterogeneous in clonal populations of many bacterial species. However, it is still unknown whether T6SS heterogeneity is genetically controlled or arises from stochastic processes and what its physiological relevance is. Here, we report that enteroaggregative E. coli (EAEC) exhibits stable phenotypic heterogeneity in T6SS expression. Under iron-limiting conditions, the Sci1 T6SS is expressed in only a subset of the population, creating distinct ON and OFF subpopulations in a reversible, heritable, and epigenetically controlled equilibrium. This heterogeneity is governed by the interplay between the iron-responsive regulator Fur- and Dam-dependent DNA methylation at the sci1 promoter. Mutations in Fur binding sites or GATC methylation motifs shift the population to homogeneous ON or OFF states, respectively. Functional analyses reveal that while ON cells mediate antibacterial activity, OFF cells buffer the population against lethal retaliatory responses from defensive T6SS⁺ competitors. Our results suggest that T6SS heterogeneity in EAEC represents a finely tuned attenuation strategy optimizing the trade-off between competitive killing and survival in hostile microbial communities. This work uncovers a novel layer of regulation in T6SS deployment and highlights phenotypic heterogeneity as an adaptive trait in interbacterial warfare.
|
|
Scooped by
mhryu@live.com
June 4, 11:10 PM
|
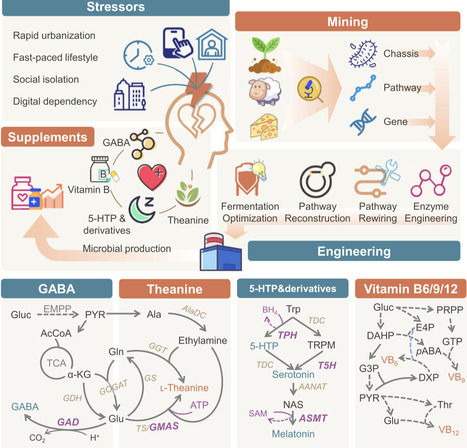
The fast-paced modern life exerts great pressure to individuals. Nutritional interventions for stress management have gained increasing attention due to its favorable safety profiles, multiple health benefits, and suitability for vulnerable populations. Microbial production of stress-relieving nutraceuticals represents a sustainable alternative to natural extraction and chemical synthesis, while meeting the growing market demand for natural-labeled and consumer-preferred products. This review provides an overview of the recent progress in engineering microorganisms to produce common stress-relieving biomolecules, such as: γ-aminobutyric acid, ʟ-theanine, 5-hydroxytryptophan and its derivatives, and vitamin B. Key bottlenecks limiting the bioproduction of each molecules and targeted strategies, including chassis selection, enzyme engineering, pathway rewiring and bioprocess regulation, are discussed.
|
|
Scooped by
mhryu@live.com
June 4, 11:02 PM
|
Photosynthetic biohybrid systems (PBSs) integrate semiconductor light harvesters with microbial metabolism to enable solar-driven chemical synthesis, yet the chemical principles governing their performance remain dispersed across two distinct architectures: wired biohybrids, which rely on photoelectrode–microbe interfaces, and wireless systems, where microbes are photosensitized by colloidal or molecular catalysts. This review examines the materials chemistry, interfacial electron transfer mechanisms, and biological constraints that define each approach. We evaluate the stability and biocompatibility of semiconductor photoelectrodes, charge transfer pathways across abiotic/biotic interfaces, microbial community dynamics, and photoelectrochemical operational parameters central to wired systems. For wireless platforms, we analyze design rules for whole-cell photosensitization, including semiconductor selection, cellular uptake, redox coupling, and mechanistic probes of electron delivery. By comparing both architectures, we identify unifying chemical principles and key bottlenecks that limit efficiency, providing a framework for the predictive design of next-generation PBSs for sustainable solar-to-chemical conversion.
|
|
Scooped by
mhryu@live.com
June 4, 10:49 PM
|
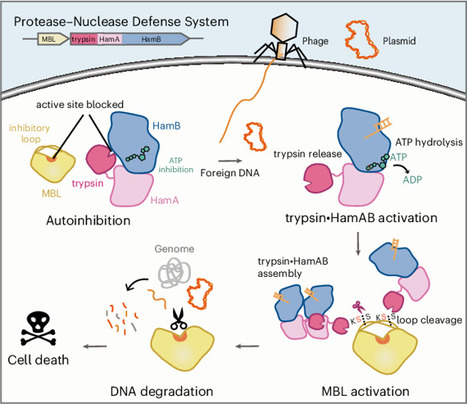
Protease-mediated activation of immune effectors is an evolutionarily conserved mechanism. This study identifies a widespread trypsin–MBL (metallo-β-lactamase) module as a core effector in diverse antiviral bacterial immune systems, such as Hachiman, AVAST and Argonaute. Focusing on the Hachiman-associated trypsin–MBL system, we show that trypsin•HamAB protease activity is inhibited by ATP, while MBL is an autoinhibited DNase with two insertion loops obstructing its catalytic site. Upon infection, trypsin•HamAB senses foreign DNA and hydrolyzes ATP, activating trypsin-like activity, which specifically cleaves MBL at the insertion loops to release repression. The activated MBL depletes DNA and arrests host cell growth. Cryo-electron microscopy structures of trypsin•HamAB–DNA reveal that DNA binding and ATP hydrolysis trigger HamAB oligomerization and trypsin-like domain release, enabling its activation. Our work elucidates a conserved immune mechanism wherein proteolytic activation of a nuclease enables robust immunity against phage while multilayered controls prevent self-toxicity, expanding the repertoire of immune processes governed by regulatory proteolysis. This study identifies and characterizes a conserved trypsin–MBL (metallo-β-lactamase) pair in bacterial immunity; upon infection, trypsin is activated through inhibitory ATP hydrolysis, subsequently activating MBL through site-specific proteolysis, which depletes DNA and restricts cell growth.
|
|
Scooped by
mhryu@live.com
June 4, 5:00 PM
|
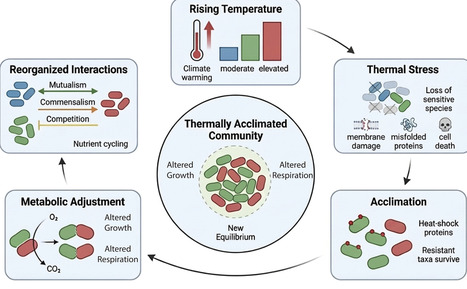
Temperature is a fundamental regulator of microbial physiology, shaping processes from growth and respiration to community interactions and ecosystem functioning. This review synthesizes recent experimental and theoretical advances that reveal how microbes respond to warming across biological scales, with a focus on short-term ecological acclimations of microbial communities. At the cellular level, rising temperature affects enzyme kinetics, membrane fluidity, and metabolic efficiency, often in non-linear ways that challenge the validity of fixed Q10-based models. At the community level, warming tends to favor thermotolerant and slow-growing taxa, while reconfiguring microbial interaction networks by shifting balances between competition, cooperation, and syntrophy. These structural changes can reduce functional redundancy and stability, yet prolonged warming may also foster the emergence of cohesive, resilient community architectures. Overall, we emphasize the need for integrative mechanistic frameworks that link thermal physiology, carbon-use efficiency, and microbial interactions to improve predictions of microbial contributions to carbon cycling under climate change.
|


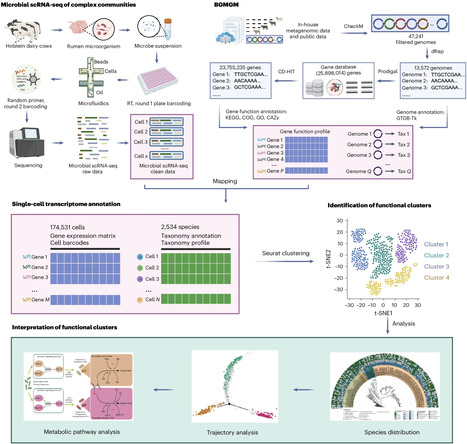




















2st, rna-seq, microbial scRNA-seq, including prokaryotic expression profiling by tagging RNA in situ and sequencing (PETRI-seq)14, microbial split-pool ligation transcriptomics (microSPLiT)15, eukaryotic bacterial droplet-based scRNA-seq (BacDrop)16 and droplet-based high-throughput single-microbe RNA-seq (smRandom-seq)17.
rumen mag database, random primer-based droplet scRNA-seq and BGMGM-based computational analysis, we develop microbiome single-cell transcriptomics (MscT) to reveal the single-cell functionalities of rumen microbiota.