 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:11 PM
|
Natural products are vital sources of medicines, and their analogues hold great promise as novel therapeutics to combat disease more effectively, including circumventing antibiotic resistance. Engineering biosynthetic pathways in microbial hosts enables efficient, cost-effective production of non-natural analogues — compounds structurally related to natural products but not necessarily derived through direct chemical modification. In this review, we highlight recent work on the in vivo production of novel natural product analogues, with an emphasis on combinatorial and precursor-directed biosynthesis, enzyme engineering, and retrobiosynthesis. We anticipate that further developments in artificial intelligence, particularly the use of machine learning models to understand enzymatic transformations and predict novel reactions, will significantly accelerate this field and drive forward its importance in drug discovery and related research.
|
|
Scooped by
mhryu@live.com
Today, 1:33 PM
|
Antibiotic resistance is a growing global health threat. Although antibiotic activity is well studied in homogeneous liquid cultures, many infections are caused by spatially structured multicellular populations where consumption of scarce nutrients establishes strong spatial variations in their abundance. These nutrient variations have long been hypothesized to help bacterial populations tolerate antibiotics, since liquid culture studies link antibiotic tolerance to metabolic activity, and thus, local nutrient availability. Here, we test this hypothesis by visualizing cell death in structured E. coli populations exposed to select nutrients and antibiotics. We find that nutrient availability acts as a bottleneck to antibiotic killing, causing death to propagate through the population as a traveling front. By integrating our measurements with biophysical theory and simulations, we establish quantitative principles that explain how collective nutrient consumption can limit the progression of this “death front,” protecting a population from a nominally deadly antibiotic dose. While increasing nutrient supply can overcome this bottleneck, in some cases, excess nutrient unexpectedly promotes the regrowth of resistant cells. Altogether, this work provides a key step toward predicting and controlling antibiotic treatment of spatially structured bacterial populations, yielding biophysical insights into collective behavior and guiding strategies for effective antibiotic stewardship. In this work, authors provide evidence that bacteria in spatially structured populations protect each other from antibiotics through collective nutrient consumption, creating ‘death fronts’ that sweep through the colony– explaining why infections often survive treatments that work in lab tests.
|
|
Scooped by
mhryu@live.com
Today, 1:08 PM
|
With antimicrobial resistance emerging as a top global health concern, new approaches to prevent and treat infections are urgently needed. Antimicrobial peptides are recognized as a promising alternative to conventional antibiotics. Considering the costly, labor-intensive, and time-consuming nature of wet lab screening, in recent years, proteomics research has increasingly relied on in silico methods to predict physicochemical, structural, and biological properties of candidate peptides. Here, we present the AMPSeek protocol to enable scientists to screen peptides and predict their potential antimicrobial activity, toxicity, and associated three-dimensional structure with a single command. The AMPSeek framework integrates AMPlify, tAMPer, and LocalColabFold, scales efficiently with increasing sample size, and produces an interactive HyperText Markup Language report to facilitate result interpretation. We provide step-by-step instructions for installing and running AMPSeek on a ten-peptide test set, along with two additional datasets (n = 20 and n = 100; mean peptide length ≈121.3) to demonstrate AMPSeek's near-linear scalability.
|
|
Scooped by
mhryu@live.com
Today, 1:01 PM
|
The transition to low-carbon energy systems requires scalable and energy-efficient routes for producing liquid biofuels that are compatible with existing fuel infrastructures. This review focuses on bio-oil production from phototrophic microorganisms, highlighting their high biomass productivity, rapid growth, and inherent capacity for carbon dioxide fixation as key advantages over conventional biofuel feedstocks. Recent progress in thermochemical conversion technologies, particularly hydrothermal liquefaction (HTL) and fast pyrolysis, is critically assessed with respect to their suitability for wet and dry algal biomass, respectively. HTL enables direct processing of high-moisture biomass while avoiding energy-intensive drying, whereas fast pyrolysis offers high bio-oil yields from lipid-rich feedstocks. In parallel, catalytic upgrading strategies, including hydrodeoxygenation and related hydroprocessing routes, are discussed as essential steps for improving bio-oil stability, heating value, and fuel compatibility. Beyond conversion technologies, innovative biological and biotechnological strategies, such as strain optimization, stress induction, co-cultivation, and synthetic biology approaches, are examined for their role in tailoring biomass composition and enhancing bio-oil precursors. The integration of microalgal cultivation with wastewater utilization is briefly considered as a supporting strategy to reduce production costs and improve overall sustainability. Overall, this review emphasizes that the effective coupling of advanced thermochemical conversion with targeted biological optimization represents the most promising pathway for scalable bio-oil production from phototrophic microorganisms, positioning algal bio-oil as a viable contributor to future low-carbon energy systems.
|
|
Scooped by
mhryu@live.com
Today, 12:50 PM
|
In Azotobacter vinelandii, the sigma factor RpoS is maintained at low levels in exponentially growing cells due to degradation mediated by the chaperone–protease complex ClpXP, while high levels are observed in the stationary phase. This study showed that degradation of RpoS by ClpXP is under the control of the Gac-Rsm signal transduction system, in which GacA, the transcriptional activator of the two-component system GacS/GacA, activates transcription of the small RNAs RsmZ1 and RsmZ2. These RNAs bind to the translational repressor protein RsmA to counteract its repressor activity on its target mRNAs. We found that in stationary-phase cells, compared with the WT, a gacA mutant exhibited lower RpoS levels due to reduced stability, while levels of the clpP and clpX mRNAs were higher. Furthermore, inactivation of the clpP or clpX genes in the gacA mutant restored the stability of RpoS, suggesting that the observed RpoS instability is due to degradation by ClpXP. We also showed that inactivation of rsmA in either the WT or the gacA mutant resulted in RpoS levels higher than in the WT in both stationary and exponential phases, while clpP and clpX transcript levels were significantly reduced. Taken together, these data reveal that in A. vinelandii, the GacA-Rsm system controls RpoS stability through RsmA, which acts as a positive regulator of ClpXP expression.
|
|
Scooped by
mhryu@live.com
Today, 12:16 PM
|
Protein structure prediction using deep learning has revolutionized protein design. Yet, our understanding of protein function remains a key limitation for designing novel proteins that perform complex biological tasks. Here, we adopt a massively-parallel, function-first approach to rationally design synthetic proteins. Using genome-scale CRISPR activation, we overexpress ~19,000 human proteins and measure their impact on precise gene editing. We identify over 800 native proteins that promote homology-directed repair. Using top candidates, we then design synthetic genome editors, Targeted Repair fUsion Editors (TruEditors), by fusing full-length proteins or smaller core domains to the Cas9 nuclease. We develop 12 unique TruEditors that improve precise gene editing in diverse cell types and at genomic loci where existing methods for precise gene editing fail. Using affinity proteomics, we show that these synthetic proteins work by coordinating with endogenous DNA repair complexes. The delivery of TruEditors via mRNA more than doubles the rate of chimeric antigen receptor (CAR) insertion into the TRAC locus of primary human T cells, enhancing CAR T cell-directed tumor cell killing, and improves precise editing in human pluripotent stem cells more than three-fold. Overall, our study demonstrates that genome-wide protein overexpression screens can guide the rational design of synthetic proteins for specific biological tasks.
|
|
Scooped by
mhryu@live.com
Today, 11:35 AM
|
Models of protein evolution are foundational to biology, underpinning essential techniques such as phylogenetic tree inference, ancestral sequence reconstruction, multiple sequence alignment, variant effect prediction, and protein design. Historically, for computational tractability, these models have relied on the simplifying -- but biologically unrealistic -- assumption that sites in a given protein evolve independently of each other. A crucial test of any evolutionary model is its ability to simulate realistic evolutionary trajectories, but the independent-sites assumption leads to simulations that poorly reflect the complexity of natural protein evolution. Here we introduce PEINT (Protein Evolution IN Time), a flexible and generalizable deep learning framework for modeling how the entire protein sequence evolves over time while incorporating complex interactions between sites. This framework enables learning realistic patterns of constrained evolutionary transitions directly from millions of protein sequences spanning diverse fold families. Furthermore, unlike classical models that require pre-aligned sequences, PEINT learns indel dynamics directly from raw, unaligned sequences, thereby eliminating potential biases from alignment errors that can lead to incorrect inference of evolutionary patterns. By capturing higher-order epistatic interactions and modeling insertion-deletion processes that classical models typically ignore, PEINT accurately reproduces key signatures of natural evolution, including conservation patterns and family-specific dynamics. When simulating evolution along phylogenetic trees, PEINT generates highly novel sequences that preserve protein function, which we validate through experimental characterization of simulated carbonic anhydrase variants that retain enzymatic activity. PEINT thus enables realistic simulation of protein evolution that explores new sequence space while respecting structural and functional constraints. This evolution-informed generative modeling framework offers a powerful new tool for advancing both phylogenetic inference and protein engineering.
|
|
Scooped by
mhryu@live.com
Today, 11:13 AM
|
Polyploidy is a ubiquitous biological phenomenon that promotes genetic innovation and adaptation. In the budding yeast Saccharomyces cerevisiae, polyploid strains are widespread in natural, industrial, and clinical niches, yet their origin is unclear. Here, we identify a novel mechanism of stepwise polyploidization that involves a Sporulate-Endoreplicate-Mate (SEM) sequence. We show that spore endoreplication during germination doubles the genome while preserving mating competence, enabling incremental increases in ploidy through mating with a non-endoreplicated spore from the same ascus. We experimentally demonstrate the transition from diploidy to triploidy and from triploidy to tetraploidy. Stepwise polyploidization thus positions triploids as central intermediates in ploidy evolution rather than evolutionary dead-ends. We further show that spores from intact asci can spontaneously undergo one or two successive SEM cycles and generate novel triploid and tetraploid strains without genetic manipulation. Natural polyploids harbor specific genomic features that are consistent with the SEM mechanism, including the prevalence of triploidy, extensive aneuploidy, pervasive heterozygosity, and a strong association with heterothallism. Together, our findings establish stepwise polyploidization through iterative SEM cycles as the predominant natural route to polyploidy in S. cerevisiae.
|
|
Scooped by
mhryu@live.com
February 20, 5:21 PM
|
Intestinal motility is a function of the enteric nervous system involving secretion of the excitatory neurotransmitter acetylcholine (ACh). Although gut commensal bacteria are key regulators of intestinal physiology, the molecular mechanisms underlying microbial influence on intestinal peristalsis and constipation remain unclear. Here we report a link between microbial nitrogen metabolism and intestinal motility regulation via ammonia production. We observed compensatory elevation of intestinal ammonia levels and urease activity in mouse models of intestinal dysmotility, induced by ACh deficiency, and in patients with constipation. Ammonia supplementation or intervention in mice with the urease-positive Lysinibacillus fusiformis isolated from patient stool, or engineered urease-expressing bacteria, effectively restored colonic ACh levels. In vitro, ammonia upregulated the expression of voltage-gated calcium channels on enteric neurons, driving Ca2+ influx to potentiate ACh secretion. Our study reveals a microbial compensatory mechanism that responds to fluctuating ACh levels in the intestine and provides microbial targets for intestinal motility disorders. Intestinal motility defects caused by lower acetylcholine levels in the gut are compensated by bacteria that generate ammonia from urea which facilitates the release of acetylcholine via upregulation of voltage-gated channels on enteric neurons.
|
|
Scooped by
mhryu@live.com
February 20, 5:07 PM
|
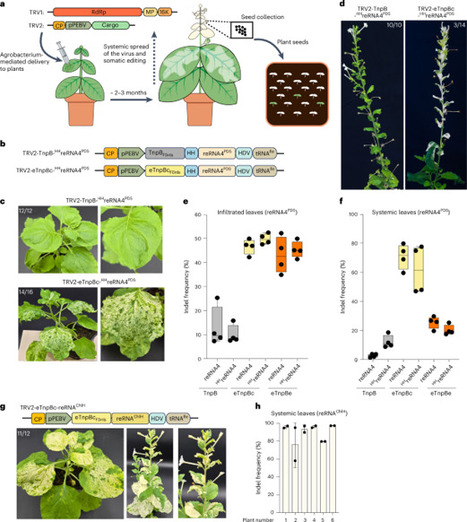
Genome editing has revolutionized plant biology research, yet the efficient delivery of editing reagents remains a challenge. Current methods are labour intensive, involving lengthy tissue culture and complex transformation and regeneration steps. Viral delivery can mitigate these issues but CRISPR–Cas nucleases exceed viral cargo limits, restricting guide RNA (gRNA) delivery into Cas9-expressing transgenic plants. This requires generating an initial Cas9 transgenic line. Furthermore, gRNAs delivered by plant viral vectors can induce somatic edits, although only a few produce heritable edits. Some engineered plant negative-strand rhabdoviruses can deliver both Cas9 and gRNA, but they face other challenges, including the need for tissue regeneration or pruning infected plants, and some rhabdoviruses can be delivered only through vector transmission. Recently, smaller editors such as TnpBs were discovered, but they are significantly less active than Cas9. Here we optimized a tobacco rattle virus-based system to deliver recently engineered, highly active ISDra2 TnpB variants. The eTnpBc variant enables effective somatic editing in systemic leaves and achieves up to 90% editing efficiency at target loci. In addition, up to 89% of offspring exhibit a mutant phenotype, with editing efficiencies reaching 100%. The design principles outlined here could promote wider use of eTnpBc for efficient, transformation- and transgene-free plant genome editing. Although genome editing has transformed plant research, delivering editing reagents remains challenging. This study reports the use of viral vectors to deliver engineered eTnpBc, achieving high levels of somatic and heritable editing in plants.
|
|
Scooped by
mhryu@live.com
February 20, 1:32 AM
|
Worsening global warming, the demand for sustainable chemicals and foods, and the societal imperative to enable healthier and longer lives are redefining the mission of bioengineering. Over the next decade, systems metabolic engineering—where systems-level understanding guides metabolic design and is empowered by synthetic biology, rapid laboratory evolution, and AI—will become an essential approach for addressing these intertwined global challenges. A central challenge is the decarbonization of chemical and materials manufacturing. Systems metabolic engineering will enable microorganisms to transform renewable biomass, industrial wastes, and even CO₂ into bulk and fine chemicals, polymers, and fuels with performance metrics that rival or surpass those of petrochemical processes. The convergence of genome-scale metabolic models, systematic identification of engineering targets and regulatory strategies, and AI-assisted pathway and enzyme design will allow cellular metabolism to be rationally designed, rewired, and optimized to maximize carbon efficiency, redox balance, and energy utilization under industrially relevant conditions. Beyond chemicals and materials, systems metabolic engineering will reshape the production of food and health molecules. Engineered microbes will provide sustainable routes to alternative proteins, functional lipids, vitamins, and structurally complex natural products that are otherwise constrained by land use, climate vulnerability, or low natural abundance. Ultimately, systems metabolic engineering will evolve from strain construction toward AI- and data-driven autonomous biomanufacturing platforms that learn, adapt, and scale, forming foundations for a low-carbon bioeconomy.
|
|
Scooped by
mhryu@live.com
February 20, 1:22 AM
|
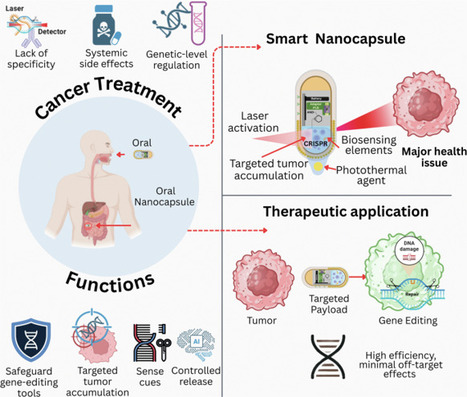
Current therapeutic techniques for cancer often lack specificity. They also cause systemic toxicity and lack genetic control. Thus, cancer ranks among the most complex and crucial global health issues. The novel concept of smart nano-capsules is discussed in this Perspective. These oral medications modify genes using CRISPR technology and integrate biosensing and laser-guided activation to enable more personalized cancer therapies. The creation of these versatile nanocapsules is driven by three objectives. First, they aim to enable controlled gene editing in the gastrointestinal tract. Second, they deliver treatments to specific target areas. Third, they detect tumors in real time. Nanocapsules equipped with biosensing components provide microenvironmental input. An external laser can trigger the release of light-absorbing agents. Moreover, these features reduce off-target effects and allow spatiotemporal precision, the enteric-coated architecture ensures oral stability. Surface functionalization enhances selective tumor accumulation. AI-guided control algorithms can manage diagnostic interpretation and activation. The CRISPR-based cancer medicines offer the potential for improved safety, specificity, and translational use in the future. Combining advanced nanotechnology, gene editing, and AI-guided control could create innovative solutions.
|
|
Scooped by
mhryu@live.com
February 20, 1:17 AM
|
Bacteroides thetaiotaomicron is a model Bacteroidota of the healthy human gut microbiota and a specialist in glycan utilization. Like other Bacteroides, B. theta has many highly regulated polysaccharide utilisation loci (PUL) that encode outer membrane (OM) TonB-dependent transporters (SusC), closely associated ''lid'' lipoproteins (SusD), and additional surface-exposed lipoproteins (SLPs) that bind and partially degrade specific glycans derived from host cells, diet, or other microbiota members. The canonical starch PUL products are thought to form a dynamic complex in the presence of starch. However, other PULs form stable complexes in the absence of substrate (recently named ''utilisomes''), with additional surface lipoproteins tightly associated with the core SusCD complex. In this study, we characterised the B. theta dextran utilisome, with a SusCDdex core and an associated glycoside hydrolase (GHdex) and surface glycan binding protein (SBGPdex). Via X-ray crystallography we solved high-resolution structures of SBGPdex in isolation and SusDdex and GHdex bound to dextran oligosaccharides. We used isothermal titration calorimetry (ITC) to quantify ligand binding of wild type and mutant SLPs. We further used single particle cryo-EM of the catalytically inactive dextran utilisome to visualize open and closed states of the complex. Three occupied dextran binding sites were observed across SusCdex, SusDdex and GHdex, with substrate observed in both open and closed states of SusDdex. 3D variability analysis showed a minority of particles in the process of SusDdex lid closure. Together our work defines commonalities and differences across utilisomes dedicated to the import of simple glycans.
|
|
|
Scooped by
mhryu@live.com
Today, 2:05 PM
|
Periodontitis is a chronic inflammatory disease driven by subgingival dysbiosis. Despite progress in characterizing microbial diversity, functional heterogeneity at the single-cell level remains poorly understood, mainly due to challenges from low microbial biomass and host contamination. Here, we present a single-cell RNA sequencing framework for profiling subgingival bacteria, enabling high-resolution analysis of these communities in health and periodontitis. Using 16 subgingival samples, we generated an atlas spanning 133,458 cells across 285 species, including 57 core active species grouped into eight functional clusters. Health-associated subpopulations specializing in adhesion and polysaccharide degradation decline in periodontitis, coinciding with hypoxia and elevated amino acid availability. The keystone pathogens T. denticola and P. gingivalis display species-specific tendencies in amino acid metabolism-related transcriptional profiles, while P. intermedia harbors a proteolytic subpopulation enriched in periodontitis. These insights deepen our understanding of periodontitis pathogenesis and inform precision diagnostics and therapeutic strategies.
|
|
Scooped by
mhryu@live.com
Today, 1:11 PM
|
Advancements in synthetic genetic circuits have enabled programmable and condition-dependent control of microbial cell growth. CRISPR-Cas9-based kill switches, genetic systems that program cells to lose viability in response to specific conditions, have recently been demonstrated for bacterial cell factories but not yet in yeast. Here, we present a foundational demonstration for a CRISPR-based kill switch in Saccharomyces cerevisiae, CRISPR KiSS. The CRISPR KiSS employs inducible CRISPR targeting essential genes to elicit growth inhibition. The activation of the KiSS system is achieved through conditional expression of a guide RNA (gRNA) upon anhydrotetracycline (ATc) induction, thereby activating CRISPR-mediated gene disruption. We demonstrate that targeting the essential genes (ERG13, PGA3, TPI1 or CDC19) leads to severe growth inhibition upon ATc induction. Still, the current set up does not allow complete killing of the cells due to system inactivation, e.g. escape from CRISPR based cutting. We studied reasons for system inactivation and substantially improved the system by simultaneous expression of two different gRNAs. Sequencing escape mutants revealed mutations in both the gRNA sequences and target genes as potential sources of system inactivation. This work highlights the potential of harnessing a CRISPR-based kill switch in S. cerevisiae. Cells expressing the system were able to escape growth inhibition through mutations and further optimization of the KiSS system is still needed for it to be used in various cell factory applications.
|
|
Scooped by
mhryu@live.com
Today, 1:05 PM
|
Phages are a valuable resource for the genetic engineering of Streptomyces antibiotic-producing bacteria. Indeed, a few integrative vectors based on phage integrase are available to insert transgenes at specific genomic loci. Chromosome conformation captures previously demonstrated that the Streptomyces linear chromosome is organized in two spatial compartments: the central compartment encompassing the most conserved and highly expressed genes in exponential phase, and the terminal compartments enriched in poorly conserved sequences including specialized metabolite biosynthetic gene clusters. This study introduces a new integrative tool based on a recently described phage, Samy, which specifically targets the terminal compartment of its native host chromosome. Samy is related to PhiC31 phage and, like the latter, encodes a serine integrase. Whereas PhiC31 targets a site generally located near the origin of replication, the Samy integration site is one of the farthest known attB sites from it. We demonstrated that the Samy integrase efficiently mediates the specific integration of a non-replicating plasmid in six Streptomyces strains from distinct clades. Bioinformatic analyses revealed that the Samy-attB site is rather conserved and located in the terminal compartment of most Streptomyces chromosomes. Finally, heterologous expression of the albonoursin biosynthetic gene cluster from the Samy-, PhiC31-, and R4-attB sites yields quantitatively equivalent levels of production, though qualitative differences were observed. Altogether, these results demonstrate that the att-int Samy system expands Streptomyces genetic engineering tools by enabling targeted integration in the terminal chromosomal compartment.
|
|
Scooped by
mhryu@live.com
Today, 12:56 PM
|
While extracellular vesicles (EVs) are established mediators of intra-species signaling, their contribution to cross-kingdom communication remains incompletely understood. Here, we investigate the EV-mediated interactions between human colon epithelial cells and both Gram-positive and Gram-negative gut bacteria. We show that bacterial EVs (BEVs) derived from Lacticaseibacillus casei, Enterococcus faecalis, and Proteus mirabilis induce distinct transcriptomic changes in Caco-2 cells depending on the bacterial species, with up to ~6,000 differentially expressed genes, including CCL20, CXCL8, or CXCL10. Transfection of BEV-derived RNA independently induces a subset of similar effects, indicating that the EV-mediated communication is partially driven by the RNA cargo. Conversely, we demonstrate that bacteria interact with Caco-2-derived EVs and miR-192-5p, which is highly abundant (~36.4-fold higher) in EVs isolated from conditioned medium compared with EVs from unconditioned medium, with modest effects on bacterial growth. Furthermore, we show that lipid-based packaging of miR-192-5p modulates its association with the bacteria. Our findings support a conceptual model in which EVs and their RNA cargo contribute to species-dependent host-microbe interactions. This study introduces a framework for understanding EVs as cross-kingdom regulators and underscores the importance of tailored, context-specific analyses for understanding the scope of EV-mediated interactions in microbiome-host homeostasis and disease.
|
|
Scooped by
mhryu@live.com
Today, 12:30 PM
|
Cyanobacteria are known for their rich secondary metabolome with a long history of research directed toward the industrial and pharmaceutical applications of their natural products. Cyanobacterial metallophores (metal-chelating molecules), however, are understudied relative to metallophores from other phyla despite evidence suggesting that genes for metallophore biosynthesis are well-represented in cyanobacterial genomes. Many of the characterized cyanobacterial metallophores are formed from hybrid biosynthetic pathways and feature mixed coordinating functional groups, leading to enhanced structural and functional diversity. The few characterized metallophore families have intriguing properties including promiscuity of metal binding, photoreactivity, and amphiphilicity that are yet to be fully explored. Research suggests that these compounds are ecologically relevant and could guide community dynamics by controlling the availability of iron, detoxifying copper, and allelopathically inhibiting certain organisms. Cyanobacterial metallophores also have potential in the fields of therapeutic design, bioremediation, technology, and agriculture. In the past five years, the number of characterized metallophores from cyanobacteria has doubled, pointing to the great promise for future discoveries.
|
|
Scooped by
mhryu@live.com
Today, 11:53 AM
|
Bacterial hypermutator strains drive rapid evolution of antibiotic resistance in chronic infections. Inspired by cancer therapy approaches that exploit synthetic lethality by targeting DNA repair deficiencies in hypermutator tumors, we tested whether pairing a conventional antibiotic with a secondary DNA-damaging agent could constrain hypermutator evolution in bacteria. Using high-throughput experimental evolution of E. coli repair-deficient strains, we evolved populations under carbapenem selection in combination with ciprofloxacin or mitomycin C. Strains lacking oxidative damage repair, double-strand break repair, or transcription-coupled repair showed significantly reduced evolvability, particularly under constant antibiotic pressure and increasing genotoxic stress. However, mismatch repair (MMR) hypermutators, the predominant clinical genotype, did not show reduced evolvability under these combination treatments. This is consistent with pathway orthogonality: MMR does not repair the structural DNA lesions induced by ciprofloxacin or mitomycin C, and the elevated mutation supply of MMR-deficient strains may allow rapid adaptation despite background DNA damage. Our findings demonstrate that combination strategies can constrain the evolvability of specific repair-deficient genotypes in vitro, but success requires matching DNA damage type to specific repair vulnerabilities. This work establishes proof of principle for genotype-directed antimicrobial strategies that exploit DNA repair vulnerabilities to constrain hypermutator evolution.
|
|
Scooped by
mhryu@live.com
Today, 11:14 AM
|
Modeling protein conformational landscapes is essential for understanding dynamics, allostery, and drug discovery, yet existing resources lack diverse conformational coverage, energetic annotations, or benchmarking standards. ProteinConformers https://zhanggroup.org/ProteinConformers provides 2.7 million geometry-optimized conformations generated with a multi-seed molecular dynamics strategy, paired with 13.7 million energy evaluations and 5.5 million similarity annotations. It delivers continuous landscapes from non-native to near-native states, benchmarking framework for multi-conformation generators, and an interactive analysis platform.
|
|
Scooped by
mhryu@live.com
Today, 10:57 AM
|
The choice between cell death (lysis) and viral dormancy (lysogeny) following bacteriophage infection serves as a founding paradigm for the emergence of cellular heterogeneity in a genetically uniform population. The determination of host fate arises through the stochastic transcription from multiple viral genomes present within each cell, but this activity remains hidden from empirical interrogation, which typically stops at the whole-cell level. Here we use parallel sequential fluorescence in situ hybridization (par-seqFISH), followed by spatial clustering of phage-encoded transcripts within each cell, to profile the transcriptional activity of individual phages during synchronized infection of E. coli by bacteriophage lambda. At the whole-cell level, transcription kinetics capture the developmental choice between lysis and lysogeny, and further demonstrate that viral replication is required for the emergence of diverging fate decisions. Zooming in to the single-phage level illuminates an individuality of viral activity during infection. We find that, while cells pursuing lysogeny display consensus activity of all inhabiting phages, lytic cells may contain phages that exhibit lysogenic activity. These findings support an earlier suggestion that consensus among coinfecting phages is required for cell dormancy. More broadly, our results highlight the need to identify how whole-cell behavior emerges from the activity of physically distinct copies of the same genetic circuit.
|
|
Scooped by
mhryu@live.com
February 20, 5:16 PM
|
Intracellular bacterial pathogens can cause high levels of morbidity and mortality in humans. Host immune responses that protect against these infections include pyroptosis, a form of lytic cell death caused by the insertion of large gasdermin (GSDM) pores into the host plasma membrane. Here we review recent advances in our understanding of how the five GSDM proteins, GSDMA–E, are activated by distinct signalling pathways. Pyroptosis can both eliminate intracellular niches and release cytosolic interleukin-1 family cytokines that further prime host immune responses against the invading pathogen. Because pyroptosis targets microbes, host-adapted intracellular pathogens have evolved strategies to efficiently subvert it. However, environmental pathogens fail to evade, making pyroptosis a potent barrier against infection. We summarize recent findings for the host-adapted bacterial pathogens Shigella flexneri, Salmonella enterica serovar Typhimurium and Mycobacterium tuberculosis, contrasted with the environmental bacteria Burkholderia thailandensis and Chromobacterium violaceum. This Review discusses recent advances in our understanding of how gasdermins are activated by distinct signalling pathways and summarizes recent findings from host-adapted bacterial pathogens contrasted with environmental bacteria.
|
|
Scooped by
mhryu@live.com
February 20, 10:14 AM
|
Protein function depends on interactions between structural domains and regulatory motifs. Yet current tools analyze these elements separately, hindering investigation of disease mutations affecting evolutionarily conserved, structurally constrained motifs. We present ProteoMapper, a computational framework integrating HMMER-based domain annotation with user-defined motif detection to quantify motif-domain spatial relationships in protein families. ProteoMapper introduces two discovery metrics: (1) positional conservation scoring, identifying motifs at identical alignment coordinates in ≥ N% of sequences (default 60%), indicating purifying selection; (2) Motif-Domain Coverage Score (MDCS), quantifying motif embedding within Pfam domains (MDCS=1: fully embedded; MDCS=0: extra-domain). The platform processes Excel-formatted alignments without programming requirements, delivering color-coded reports with conserved motif positions, domain boundaries, and MDCS values. Parallel execution of sequence batches enables rapid analysis (8 motifs were searched in 150 sequences with complete Pfam scanning in <6 seconds on standard hardware). Validation across three protein families confirmed technical accuracy and biological insight. In PLATZ transcription factors (24 proteins), domain predictions achieved 0.94 mean intersection-over-union versus published annotations, exactly reproducing 22 of 23 reported spans. In Arabidopsis ERD6-like sugar transporters (17 proteins), MDCS analysis revealed canonical PROSITE signatures PS00216 and PS00217 are equally domain-embedded (MDCS=1.0) but evolutionarily divergent. PS00217 shows positional conservation (58.8% of sequences) while PS00216 exhibits dispersal, suggesting subfunctionalization. In tomato actin-depolymerizing factors (11 proteins), domain detection achieved 100% sensitivity with >93% positional concordance. ProteoMapper enables hypothesis-driven investigation of evolutionary constraints, regulatory mechanisms, and variant effect prediction in biomedical and functional proteomics. https://github.com/sifullah0/ProteoMapper.
|
|
Scooped by
mhryu@live.com
February 20, 1:26 AM
|
RNA design aims to find a sequence that can fold into a target secondary structure. It can create artificial RNA molecules for specific functions, with wide applications in medicine. It is computationally challenging due to two levels of combinatorial explosion: the exponentially large design space and the exponentially many competing structures per design. Popular methods such as local search cannot keep up with these combinatorial explosions. We instead employ two techniques from machine learning, continuous optimization and Monte-Carlo sampling. We start from a distribution over all valid sequences, and use gradient descent to improve the expectation of an arbitrary objective function. We define novel coupled-variable distributions to model the correlation between nucleotides. We then use sampling to approximate the objective, estimate the gradient, and select the final candidate. Our work consistently outperforms state-of-the-art methods in key metrics including Boltzmann probability and ensemble defect, especially on long and hard-to-design structures. RNA design aims to find a sequence that folds into a target structure. Here, the authors formulate it as a continuous optimization over a coupled-variable distribution and leverage sampling to optimize arbitrary objectives. On Eterna100, it outperforms state-of-the-art methods across key metrics.
|
|
Scooped by
mhryu@live.com
February 20, 1:20 AM
|
Lignin represents a promising and sustainable feedstock for the production of high-value products. However, its heterogeneity and recalcitrance pose a big challenge for efficient depolymerization and upcycling. This review highlights recent advances in microbial lignin valorization, focusing on three key steps: lignin depolymerization, metabolism of lignin-derived aromatic compounds, and valorization to target products. Recent progress in lignin depolymerization is enabling the discovery and optimization of more efficient and broadly specific ligninolytic enzymes, and highlights the critical role of auxiliary enzymes and quinone redox cycling in supporting ligninolytic activity. New catabolic mechanisms, transport systems, and transcriptional regulation networks for both dimeric and monomeric lignin-derived substrates expand our understanding of biological funneling pathways, and they offer valuable tools for designing more efficient microbial biocatalysts and biosensors. Emerging metabolic engineering and adaptive laboratory evolution strategies for creating robust microbial chassis capable of producing diverse value-added products from lignin-derived feedstocks are discussed.
|































1str